Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
Gastric cancer (GC) is common but often diagnosed late. Recent advances in chemotherapy, targeted therapy, and immunotherapy offer promising treatments. Perioperative chemotherapy is now the standard for resectable gastric cancer. Progress has also been made in treating metastatic disease using targeted immunotherapies. Molecular biomarkers such as PD-L1, MSI, and HER2 guide personalized treatment approaches.
- novel oncogene targets
- gastric cancer
- immune biomarker
1. Introduction
Gastric cancer (GC) is a significant global health concern, ranking as the fifth most common cancer and the fourth leading cause of cancer-related deaths worldwide in 2020, according to the Global Cancer Observatory (GLOBOCAN) [1]. Its incidence is particularly pronounced in East Asia and Eastern Europe [1]. More than 95% of GC cases are adenocarcinomas. Detecting early GC poses a significant challenge due to the delayed emergence of clinical symptoms, which precludes surgical intervention. Considering poor survival rates among GC patients worldwide, especially those with metastatic disease, there is a tremendous unmet need to uncover novel therapeutic approaches to improve patient outcomes [2].
The mechanisms behind GC pathogenesis remain unclear and involve a complex interplay of environmental and genetic factors [2]. Dysregulation of numerous genes and pathways plays a pivotal role during gastric carcinogenesis. The recent understanding of molecular signaling pathways is contributing to our knowledge of the tumor’s pathogenesis and provides valuable insights into the potential of targeted therapies [3]. Consequently, new molecular classifications have been proposed to better understand GC and its subtypes [4,5].
2. Histological and Molecular Classification
Various classification systems exist for gastric cancer as depicted in Figure 1 [6]. The Lauren classification, established in 1965, divides it into intestinal and diffuse types based on histopathology [6]. The 2010 World Health Organization (WHO) classification, revised in 2019, organizes gastric cancer into distinct subtypes of adenocarcinomas, including papillary, tubular, mucinous, poorly cohesive, and less common forms of gastric tumors [7].
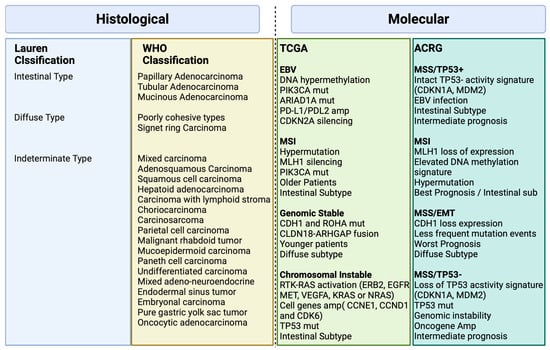
Figure 1. Histological and Molecular Classification in Gastric Cancer Biorender agreement number: JO25V7ORTC.
There was a need for a new molecular classification system since histological classifications do not take into account the molecular profiling features in gastric cancer [4]. The Cancer Genome Atlas (TCGA) [4] proposed a classification that identifies dysregulated pathways and potential gene mutations in four distinct molecular subtype groups. The first subtype comprises Epstein–Barr virus (EBV)-positive tumors (9%) characterized by high levels of DNA hypermethylation. The second subtype includes microsatellite instability (MSI) tumors (22%) with a hypermutated genome, DNA hypermethylation, and MLH1 silencing. The third subtype consists of genomically stable (GS) tumors (20%) exhibiting a low mutation burden and displaying more aggressive disease traits. Over 70% of these tumors demonstrate diffuse histology and harbor somatic CDH1 mutations and Claudin 18.2 rearrangements. The final subtype is chromosomal instability (CIN) tumors (50%) which are associated with intestinal histology and exhibit high somatic copy number aberrations, common TP53 mutations, and amplifications of the RAS receptor tyrosine kinase pathway. In the CIN subtype, most affected genes are vascular endothelial growth factor (VEGF), epidermal growth factor receptor (EGFR) (10%), Human epidermal growth factor receptor (ERBB2) (24%), ERBB3 (8%), FGFR2 (8%), and c-Met (8%) (Figure 2).
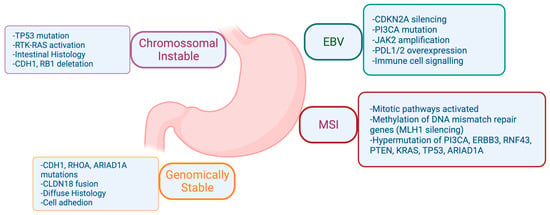
Figure 2. Comprehensive Molecular Characterization of Gastric Adenocarcinoma according to The Cancer Genome Atlas (TCGA). Biorender agreement number CU25Y0RJS4.
The Asian Cancer Research Group (ACRG) [5] conducted a study that built on the TCGA analysis, incorporating additional molecular analysis, and correlating the results with clinical outcomes data. They identified four distinct subtypes of gastric cancer: MSI-High (23% of patients) with the best prognosis, typically diagnosed in early-stage cancer; microsatellite stable (MSS) and epithelial–mesenchymal transition (EMT) (15% of patients) with the poorest overall prognosis, associated with diffuse histology (80%) and advanced clinical stages (III/IV); MSS/TP53 intact (26% of patients) with the second-best prognosis and a high rate of EBV infection (15% of patients) and finally, MSS/TP53 loss (36% of patients) with the highest rate of TP53 mutations and a less favorable prognosis than MSI and MSS/epithelial/TP53 intact, but better than MSS/EMT [5].
Figure 1 illustrates the major features and genomic alterations associated with each GC subtype according to Histological (Lauren and WHO) and Molecular (TCGA and ACRG studies) classification.
3. Risk Factors and Molecular Mechanisms
Hereditary syndromes and familial GC, characterized by specific family history criteria, occur in less than 10% of cases [8]. Ninety percent of GC is non-hereditary and associated with several factors, occurring sporadically. Non-cardia tumors are often associated with Helicobacter pylori infection [9], with chronic infection leading to chronic gastritis, and continued inflammation leading to damage to the stomach lining, ulcers, and gastric atrophy [10]. H. pylori inflammation is primarily caused by pathogenic factors like cytotoxin-related gene A (CagA), vacuolar cytotoxin A (VacA), and the Cag IV secretion system, encoded by specific genes within the CagA pathogenicity island (PAI) DNA insertion element [11]. These virulence factors are released, activating signaling pathways such as NF-κB, p53 and JAK-STAT. A detailed discussion of these pathways will be discussed in Section 4. The TP53 gene exhibited increased activity in individuals with H. pylori infection, as evidenced by both the TCGA database and its RNA sequencing data [4]. A total of 33 signaling pathways and 10 biological processes demonstrated a significant positive correlation with H. pylori infection [4].
Both cardia and non-cardia tumors share risk factors such as alcohol consumption and tobacco smoking [12]. Tumors situated in the cardia region are, on the other hand, more likely to be linked with gastroesophageal reflux and EBV infection [13]. EBV infection may lead to increased DNA hypermethylation, frequent PIK3CA mutations, and overexpression of JAK2 [13]. Furthermore, programmed cell death protein ligand 1 (PD-L1), expressed in certain tumor cells, inhibits immune responses by binding to PD-1 receptors on T lymphocytes. Elevated levels of PD-L1, whether on tumor cells or infiltrating lymphocytes, correlate with improved response rates and overall survival (OS) when treated with immunotherapy in different tumors [14]. A systematic review estimated a PD-L1 positivity rate of approximately 55% in EBV-associated gastric cancer, but with significant variability among studies, likely linked to histologic variations. Specifically, GC-LS (lymphoid stroma) tumors, often associated with EBV, exhibit a PD-L1 positivity rate exceeding 80%. In EBV-GC, higher PD-L1 expression is observed, suggesting the potential of EBV positivity as a marker for immunotherapy selection [14].
The key molecular mechanisms involved in gastric cancer development caused by EBV will be discussed further. See discussions in Section 4.
4. Molecular Mechanisms and Signaling Pathways in Gastric Cancer
In 2014, the TCGA Research Network conducted a groundbreaking study that represented one of the most comprehensive molecular characterizations of gastric adenocarcinoma [4]. The study involved the evaluation of 295 primary gastric adenocarcinomas, employing a comprehensive approach encompassing six molecular platforms: array-based somatic copy number analysis, whole-exome sequencing, array-based DNA methylation profiling, messenger ribonucleic acid (RNA) sequencing, microRNA (miRNA) sequencing, and reverse-phase protein array (RPPA). Additionally, MSI testing was performed on all tumors, adding a crucial dimension to the molecular analysis. Through this rigorous examination, TCGA was able to categorize gastric cancer into four distinct subtypes, its unique pathways and genomic features involved in gastric adenocarcinoma (Figure 2).
However, the tumorigenesis process entails a diverse array of signaling pathways, beyond those spotlighted in Figure 2, as depicted in Figure 3. Within the context of gastric cancer, we delve into these specific signaling pathways that influence invasion and tumor progression. Furthermore, the subsequent section will thoroughly explore signaling pathways, their frequencies, their prognostic implications, and their significance in both gastric cancer research and treatment.
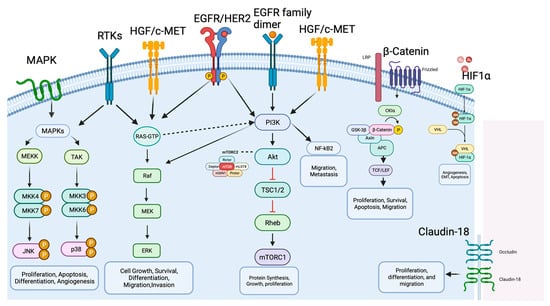
Figure 3. Molecular mechanisms and signaling pathways in gastric cancer. The figure illustrates the main molecular pathways and key factors involved in GC development and progression, including MAPK, HER2, PI3K/AKT/mTOR, HGF/c-Met, p53, Wnt/β-catenin, NF-κB and PD-1/PD-L1 and CTL4. The arrows represent activation signals and there are no difference between the arrows type (dashed versus solid). Biorender agreement number: VO25V7OL5G.
4.1. MAPK Signaling Pathway
The mitogen-activated protein kinase (MAPK) signaling pathway represents an extensive group of serine/threonine protein kinases crucial for determining how cells respond to various external triggers [14]. In GC, the RAS/RAF/MAPK and PI3K/AKT/mTOR signaling pathways are notable among the highly impaired pathways [15].
The MAPK signaling pathway is composed of five interconnected cascades: extracellular signal-related kinases (ERK), specifically ERK1/2; Jun amino-terminal kinases (JNK), encompassing SAPK/JNK1, JNK2, and JNK; and p38-MAPK, which involves p38α, p38β, p38γ, and p38δ, along with ERK5 and ERK3 [16]. In GC, the MAPK signaling pathway is responsible for the processes of invasion and metastasis, such processes involve motility and cell adhesion and disintegration of focal adhesions triggered by the epidermal growth factor receptor (EGFR), which is regulated by activating the ERK/MAPK pathways [16].
The JNK subgroup within the MAPKs family responds to stress signals, a trigger for cell proliferation, apoptosis, or transformation [17]. A study involving mice highlighted JNK’s association with tumor initiation and progression, suggesting it as a promising target for GC prevention [18].
Within cancer, the p38-MAPK pathway exhibits intricate regulation. Numerous studies have indicated that p38 serves as an oncogenic factor, playing a pivotal role in pathological events linked to tumor progression, such as inflammation, invasion, angiogenesis, and chemotherapy resistance in GC [19].
Moreover, miRNAs, like miR-302b, MiR-20a, miR-21, and miR-106a possess regulatory abilities in various cellular pathways. These miRNAs have been recognized as controllers of MAPKs, modulating the proliferation, survival, and metastasis of gastric cancer cells [20]. Through experiments conducted on GC cell lines, it was observed that miR-491-5p effectively suppressed cell migration and proliferation while promoting apoptosis. Initially identified as an inhibitor of the antiapoptotic factor BCL-XL, miR-491-5p has been demonstrated to inhibit ERK1/2 and AKT signaling pathways [20].
4.2. HER2 Signaling Pathway
Human epidermal growth factor receptor 2 (HER2 or erb2) is an oncogene encoded by erb2 on chromosome 17 and belongs to the EGFR family of receptor tyrosine kinases [21]. In HER2-induced cancers, particularly HER2-positive tumors, the HER2/HER3 dimer plays a crucial role in tumorigenesis and tumor maintenance as the most potent heterodimer within the EGFR family. Hence, directing efforts towards inhibiting the association of HER2 with other EGFR family counterparts, especially HER3, represents a viable therapeutic strategy for addressing HER2-positive tumors [22].
The oncogenic Impacts of elevated HER2 levels arise from the spontaneous formation of receptor homodimers or heterodimers with other members of the EGFR family. This activation initiates downstream signaling cascades, including the PI3K/AKT/mTOR and MAPK/ERK1/2 pathways. The activation of these pathways fosters diverse cellular processes vital for tumor progression, such as cell proliferation, differentiation, survival, angiogenesis, and metastasis [21,22].
The phase III Trastuzumab for Gastric Cancer (ToGA) trial reported the incidence of HER2-positive gastric cancer to be 22% [23], but it can vary in the literature. The PI3K/AKT/mTOR signaling pathway is implicated in the molecular mechanisms associated with HER2-driven tumorigenesis [24]. Therefore, targeting HER2 and its downstream signaling pathways holds potential as a therapeutic strategy for HER2-positive tumors [21].
4.3. PI3K/AKT/mTOR Signaling Pathway
The PI3K/AKT/mTOR pathway is frequently dysregulated in GC [25]. PIK3CA mutations are present in approximately 9–12% patients with non-hypermutated tumors and 32% patients with hypermutated tumors [25,26]. A range of mutations within the PIK3CA gene and amplification of receptor tyrosine kinase genes such as EGFR and HER2 were detected in the analyzed cases of gastric cancer [27]. While some studies have not found a significant association between PIK3CA mutations and clinical outcomes, the amplification of PIK3CA is closely linked to the advancement of tumors, prognostic outcomes, and the emergence of drug resistance in GC [27].
In GC, the PI3K/AKT/mTOR signaling pathway assumes a pivotal role, propelling tumor progression via a diverse range of mechanisms such as impeding apoptosis, fostering drug resistance, facilitating metastasis, and promoting angiogenesis [28]. Perturbations in this pathway notably play a key part in resistance to HER2-targeted therapies and the development of chemoresistance in breast cancer [29]. Given the substantial engagement of the PI3K/AKT/mTOR signaling pathway in the progression of GC, aiming interventions at this intricate signaling axis represents a promising yet challenging therapeutic strategy for effective GC treatment.
4.4. HGF/c-MET Signaling Pathway
The mesenchymal epidermal transition factor (c-MET), which is encoded by the proto-oncogene MET, is a transmembrane receptor expressed on the surface of epithelial and endothelial cells [30]. c-MET is a receptor tyrosine kinase belonging to the MET (MNNG HOS transforming gene) proteins. Its specific ligand is hepatocyte growth factor (HGF). When HGF binds to c-MET, it activates the canonical pathway by causing c-MET to form homodimers and phosphorylate its intracellular kinase domains [30]. The HGF/c-MET pathway plays crucial roles in normal cellular processes, but its abnormal activation is strongly linked to tumor invasion and metastasis in various epithelial cancers. This aberrant activation can occur through multiple mechanisms including gene amplification, activating mutations, changes in gene expression, overexpression of c-MET or HGF, increased stimulation by self-produced or neighboring HGF molecules, the interaction with other active receptors on the cell surface, and disruptions caused by environmental factors such as low oxygen levels and inflammation [31]. While gene amplification of MET is infrequent in gastric cancer p”Iie’ts (4–10%) [32], an overexpression of the c-MET protein has been identified in a significant proportion of cases, ranging from 18% to 82% [33]. The complexity of the MET signaling pathway, the lack of consensus and poor biomarker determination as well as the diverse resistance mechanisms (crosstalk, novel bypass mutations, upregulated gene amplification), resulted in the limitation of clinical efficacy of MET inhibition [34].
4.5. Wnt/β-Catenin Signaling Pathway
The Wnt/β-catenin signaling pathway is involved in cell proliferation, migration, and cell death, and is important for the development and homeostasis of various tissues [35]. Abnormal activation of the Wnt/β-catenin pathway, crucial in the malignant transformation and invasion of GC, is brought about by mutations in various key components of the standard Wnt signaling [36]. This pathway becomes activated when Wnt ligands connect with Frizzled (FZD) and LRP5/6 receptors, initiating the recruitment of intracellular proteins like Disheveled (DVL) and Axin. This activation inhibits the phosphorylation of β-catenin and ensures its stability. Accumulation of β-catenin within the cytoplasm permits its dissociation from degradation complexes, facilitating its entry into the nucleus. Inside the nucleus, β-catenin collaborates with T-cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors to activate genes responsive to Wnt signaling [37]. Disrupted activation of the Wnt/β-catenin pathway, frequently resulting from mutations in its constituents, plays a role in the malignant transformation and invasion of GC [38].
Furthermore, this pathway plays a role in governing the infiltration of T cells within the tumor microenvironment and influences the response to programmed cell death protein 1 (PD-1) antibodies. Additionally, activated Wnt/β-catenin signaling in GC brings about further mechanisms, such as the adjustment of immunoregulatory factors like CCL28 (Mucosae-associated epithelial chemokine) and the interplay between β-catenin and E-cadherin [39]. An intriguing finding is the unexpected antitumor effect observed upon CCL28 blockade, which effectively hinders the infiltration of Treg cells. This discovery holds significant importance as it introduces a novel concept for the immunotherapy of gastric cancer, potentially leading to innovative diagnostic approaches and therapeutic interventions in the foreseeable future [40].
4.6. FGF/FGFR Signaling Pathway
The fibroblast growth factor receptors (FGFRs) represent a group of transmembrane proteins found across various cell types [41]. This family encompasses four distinct members, including FGFR1, FGFR2, FGFR3, and FGFR4. In the context of GC, common FGFR alterations include mutations in FGFR1, amplification of FGFR2, and rearrangements in FGFR3 [41]. The activation of FGFRs occurs upon binding of fibroblast growth factors (FGFs), leading to the phosphorylation of the intracellular tyrosine kinase domain and the initiation of several critical cellular pathways [42]. These pathways encompass the RAS/MAPK, PIK3CA/AKT/mTOR, and Janus kinase (JAK) pathways, which can influence various processes like angiogenesis, cell mitosis, differentiation, proliferation, and invasion [41,42].
The prevalence of FGFR abnormalities in GC ranges from 4.1% to 7% [43,44], with amplifications being the most frequently observed, followed by rearrangements and mutations. Cancers exhibiting co-alterations in FGFR2, c-Jun, and YAP1 have shown associations with poorer clinical outcomes [45]. Moreover, FGFR has emerged as a potential prognostic biomarker and therapeutic target in inhibiting the development of gastric tumors [46,47,48].
4.7. HIF-1α Signaling Pathway
HIF-1α plays a crucial role in cellular adaptation to low oxygen levels. When cells experience hypoxia, HIF-1α expression increases, and the activity of hydroxylases, which normally inhibit HIF-1α, is suppressed due to the absence of oxygen. This leads to the activation of HIF-1α, which then moves to the cell nucleus. Once in the nucleus, HIF-1α functions as a transcription factor, influencing the regulation of various target genes involved in metabolism, inflammation, vascular homeostasis, and tumor formation [49]. The HIF-1α signaling pathway is believed to play a significant role in advancing the progression of GC by facilitating tumor cell growth, promoting angiogenesis, inducing EMT, fostering resistance to therapy, and inhibiting programmed cell death. Increased expression of HIF-1α in GC patients may serve as an indicator of unfavorable OS outcomes [50].
4.8. Claudin 18.2 Signaling Pathway
Claudins are transmembrane proteins which maintain the tight junction between cells which form a paracellular barrier to control the flow of molecules between cells. Claudin-18 (CLDN18.2) has been linked with prognosis in GC [51]. Aberrant expression of CLDN18.2 is commonly observed in the initiation and progression of various malignancies. In the context of gastric epithelial tissue undergoing malignant transformation, disruptions in cellular polarity result in the exposure of CLDN18.2 epitopes on the cell surface, leading to its highly selective and stable expression in specific tumor tissues [52]. The CLDN18.2 protein plays a role in tumor cell proliferation, differentiation, and migration. The stomach-specific isoform, CLDN18 isoform 2 (CLDN18.2) [53], is emerging as a promising treatment target because of high expression in GC cells, including targeting via adoptive T-cell strategies. According to a meta-analysis the expression of CLDN18.2 was detected in approximately 34.2% of a combined population of 2055 patients [54].
4.9. NF-κB Signaling Pathway
The NF-κB family of transcription factors, including RelA, RelB, c-Rel, NF-κB1 (p50), and NF-κB2 (p52), regulate the expression of various genes involved in cell survival, apoptosis, and inflammation [55]. Activation of the canonical NF-κB pathway occurs when receptors such as TNFRs, TLRs, and IL-1R stimulate the IκB kinase (IKK) complex, leading to the degradation of IκBα. This allows NF-κB to translocate to the nucleus and activate downstream target genes, contributing to GC progression and metastasis [56]. Aberrant NF-κB signaling is associated with anti-apoptotic factor in GC [57]. The inhibition of NF-κB signaling shows challenging potential in inducing apoptosis, cell cycle arrest, and enhancing the efficacy of chemotherapy. The PI3K/AKT pathway regulates the NF-kB cascade, leading to NF-kB activation promoting cell invasion and migration [57].
4.10. TGF-β Signaling Pathway
Transforming growth piter-β (TGF-β) is an f of polypeptides involved in various physiological processes, including embryonic growth and inflammation regulation [58]. TGF-β1, the most abundant form, has complex roles in cell growth regulation and is linked to tumor development. In GC, TGF-β1 influences clinical features and patient prognosis, sometimes inhibiting cell growth but also promoting tumor progression. TGF-β signaling induces an epithelial–mesenchymal transition (EMT) in GC through the interaction with the AMPK pathway. Targeting Smad3 phosphorylation in TGF-β signaling may offer a potential therapeutic strategy for GC [58,59,60].
4.11. P53 Signaling Pathway
P53 holds a central role in overseeing DNA repair and governing the cell cycle, apoptosis, and differentiation by means of interactions between DNA and proteins, as well as interactions between proteins themselves [61]. It holds significant importance in averting the buildup of potentially cancerous or flawed cells by inducing aging, promoting cell apoptosis, and facilitating DNA repair. Elevated expression levels of p53 have been observed in GC patients, with a TP53 gene mutation rate of approximately 30% across all GC patients. However, this mutation rate may vary among patients with different GC subtypes and etiologies [62]. The presence of H. pylori infection has been linked to the accumulation of TP53 gene mutations, reported in approximately 50% of gastric tumors [63]. This occurs due to genotoxic stress, where p53 activates signaling pathways that result in temporary cell cycle arrest, allowing for DNA repair processes to take place. Inactivation of p53 promotes genomic instability, a characteristic feature of cancer, further underscoring its significance in GC development [63].
4.12. STAT3 Signaling Pathway
Signal transducers and activators of transcription 3 (STAT3) is a hyperactivated oncogene found in various cancers, including GC. It is activated by cytokine binding to transmembrane receptors, leading to dimerization and transphosphorylation of JAKs [64]. Upon activation, STAT3 relocates to the nucleus, where it modulates the expression of genes linked to the proliferation, invasion, and resistance to chemotherapy in cancer cells [65]. Both STAT3 and Survivin hold promise as potential indicators for prognosis and targets for therapeutic interventions in GC [66]. The JAK2/STAT3 pathway is also implicated in GC metastasis and epithelial–mesenchymal transition. MicroRNAs, long non-coding RNAs, and circular RNAs have been identified as regulators of STAT3 in GC, potentially influencing its expression levels and contributing to chemoresistance and tumor progression [67].
4.13. PDL-1/PD-1/CTLA-4
Cancer growth and progression can also be related to the suppression of the immune system. Immune checkpoints assume a pivotal function in this process [68]. These checkpoints can either stimulate or inhibit immune cell activity, maintaining a balance in the immune response. Some inhibitory checkpoints like PD-1, CTLA-4, TIM-3, LAG-3, and TIGIT are expressed on T cells and regulate immune signaling pathways, preventing excessive immune damage [69]. In tumor cells, these checkpoints are upregulated during tumor progression, suppressing the body’s ability to mount an effective anti-tumor immune response, and allowing the tumor to evade immune attack. Therefore, targeting immune checkpoints has become a vital approach in cancer immunotherapy [68].
One of the well-studied pathways is the PD-1/PD-L1 signaling, where PD-L1 and PD-L2 proteins on cancer cells interact with PD-1 on T cells, reducing T cell activity and promoting cancer cell survival [69]. Similarly, the CD28/CTLA-4/B7 pathway involves proteins like CD28 and CTLA-4 on T cells and B7-1/2 on antigen-presenting cells. The binding of B7-1/2 to CTLA-4 inhibits T-cell activity, preventing the killing of tumor cells [69]. Blocking these interactions using immune checkpoint inhibitors, like anti-CTLA-4 antibodies, can activate T cells and enhance their ability to target and eliminate cancer cells [69]. Other pathways involving TIM-3, LAG-3, and TIGIT also contribute to immune tolerance and dysfunction, making them potential targets for cancer therapy as well [70]. Among these, PD-1/PD-L1 signaling has received extensive attention as a diagnostic and prognostic biomarker and a therapeutic target for GC treatment.
4.14. MSI High
In normal cells, the DNA mismatch repair (MMR) system ensures genomic accuracy during DNA replication by recognizing and repairing genetic mismatches. However, in MSI tumor cells with deficient MMR (dMMR), microsatellite DNA mismatches accumulate, leading to mutations in various genomic regions, primarily involving key MMR components like MLH1, MSH2, MSH6, and PMS2/1 [71]. Disruption of MMR proteins can occur due to mutations within the coding region, promoter methylation, or chromosomal rearrangements, ultimately resulting in loss of heterozygosity.
In gastric cancer, MSI prevalence varies with tumor stage, being highest in node-negative disease (up to 20%) and lowest in metastatic disease (<5%), with a higher occurrence in the intestinal type [72]. While hereditary Lynch syndrome is a rare cause (~1.5%) of GC, epigenetic silencing of hMLH1 through promoter hypermethylation is a leading factor in MMR deficiency for both sporadic and familial MSI GCs [73].
Extensive research has advanced our understanding of the molecular landscape of MSI GCs. The whole genome sequencing performed by the TCGA group has elucidated genes significantly mutated in MSI GCs, encompassing crucial processes like cell cycle progression and regulation (e.g., TP53, IGFIIR, and TCF4), DNA integrity maintenance (e.g., hMSH6, hMSH3, MED1, RAD50, BLM, ATR, and MRE11), chromatin remodeling, cell death (e.g., RIZ, BAX, CASPASE5, FAS, BCL10, and APAF1), transcription regulation, and signal transduction [4]. Additionally, mutations affecting major histocompatibility complex class I genes, such as B2M and HLA-B, contribute to a loss of HLA class 1 complex expression, reducing antigen presentation and enabling an immune-surveillance escape.
Moreover, specific mutations have been identified in genes like EGFR, KRAS, PIK3CA, and MLK3, with varying prevalence across MSI cases [74]. The association between KRAS mutations and MSI status has been firmly supported by numerous studies (30% in MSI tumors) [74]. Other genes frequently mutated in MSI GC, such as ARID1A and RNF43, further contribute to the intricate molecular landscape.
This entry is adapted from the peer-reviewed paper 10.3390/cancers15205075
This entry is offline, you can click here to edit this entry!