The epithelial-mesenchymal transition (EMT) is a complicated biological process in which cells with epithelial phenotype are transformed into mesenchymal cells with loss of cell polarity and cell–cell adhesion and gain of the ability to migrate. EMT and the reverse mesenchymal-epithelial transitions (METs) are present during cancer progression and metastasis. Using the dynamic switch between EMT and MET, tumour cells can migrate to neighbouring organs or metastasize in the distance and develop resistance to traditional chemotherapy and targeted drug treatments. Growing evidence shows that reversing or inhibiting EMT may be an advantageous approach for suppressing the migration of tumour cells or distant metastasis. Among different levels of modulation of EMT, alternative splicing (AS) plays an important role.
- epithelial-mesenchymal transitions
- alternative splicing
- cancer
1. Introduction
2. The Epithelial-Mesenchymal Transition
2.1. Mechanism of EMT
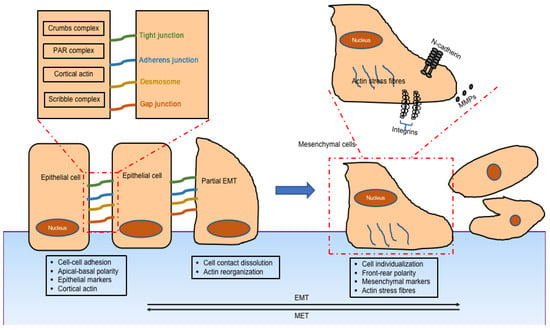
2.2. Classification of EMT
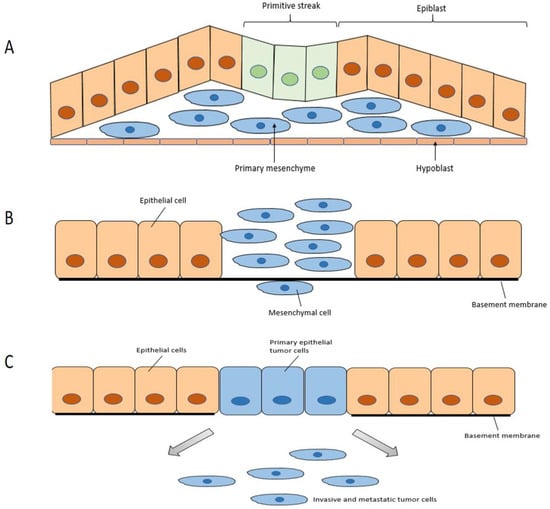
2.3. Regulation of EMT
3. Alternative Splicing
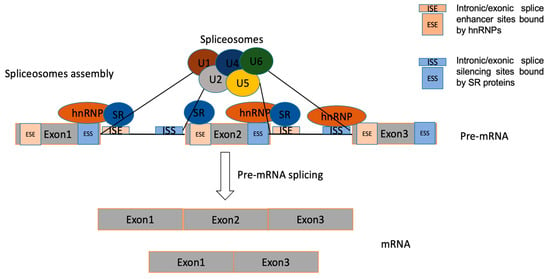
3.1. Splicing
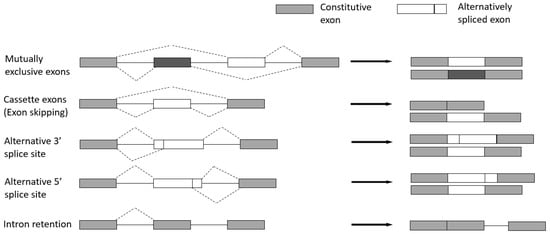
3.2. Basic Modes of Alternative Splicing
-
Cassette exons (Exon skipping)—the most prevalent pattern in which exons are spliced out of the gene or retained in the transcript.
-
Mutually exclusive exons—only one of two consecutive exons is retained in the mature transcript.
-
Alternative 3′ splice site—when the splice junction at the 3′ end is changed.
-
Alternative 5′ splice site—when the splice junction at the 5′ end is changed. Both alternative 3′ and 5′ splice sites can cause changes in the coding sequence.
-
Intron retention—when an intron is retained in the final transcript.
3.3. Regulation of Alternative Splicing
The regulation of AS is a highly dynamic combinatorial process that relies on complex coordination between intracellular and post-transcriptional processes [23]. Regulation of AS can be triggered by the interaction of multiple RNA-binding proteins (RBPs), which bind to cis-regulatory elements around the splice sites resulting in the utilization of the regulated splice site being enhanced or repressed [20]. The cis-elements, which can be located in either exons or introns, are bound by regulatory proteins to enhance or silence splicing of adjacent regulated exons, to therefore determine whether the mature transcript includes or skips certain exons [24]. Splicing regulatory elements can be divided into four categories: exonic/intronic splicing enhancer/silencers [25]. Precise control of AS is vital for normal cells, as aberrant expression of splicing is a common cause of diseases including cancer. The trans-acting splicing factors belong mostly to two large families—SR proteins, which normally bind to splicing enhancers and promote spliceosomes assembly, and hnRNPs opposing SR protein function, which generally bind to splicing silencers to inhibit exon recognition and promote exon skipping [26][27].
4. Manipulation of Alternative Splicing in EMT as a Potential Therapy for Cancer
4.1. Switching Specific Alternative Splicing Patterns in EMT
4.1.1. Fibroblast Growth Factor Receptor 2 (FGFR2)
4.1.2. Recepteur d’Origine Nantais (RON, MST1R)
4.1.3. CD44
4.1.4. Catenin Delta 1 (CTNND1)
4.1.5. Other Genes Spliced Differently during EMT
ARHGEF11 (Rho guanine nucleotide exchange factor 11) is the guanine nucleotide exchange factor (GEF) for the RhoA small GTPase protein. It has been revealed to promote tumour metastasis in glioblastoma and ovarian carcinoma and promotes proliferation and EMT of hepatocellular carcinoma by activating β-catenin [52].
4.2. Targeting Splicing Regulators Involved in EMT
4.2.1. Epithelial Splicing Regulatory Proteins (ESRPs)
4.2.2. RNA-Binding Fox Protein 2 (RBFOX2)
4.2.3. Other Splicing Factors Involved in EMT
5. Conclusions
This entry is adapted from the peer-reviewed paper 10.3390/genes14112001
References
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428.
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45.
- Christofori, G. New signals from the invasive front. Nature 2006, 441, 444–450.
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110.
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437.
- Biamonti, G.; Bonomi, S.; Gallo, S.; Ghigna, C. Making alternative splicing decisions during epithelial-to-mesenchymal transition (EMT). Cell. Mol. Life Sci. 2012, 69, 2515–2526.
- Nieto, M.A. Epithelial Plasticity: A Common Theme in Embryonic and Cancer Cells. Science 2013, 342, 1234850.
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196.
- Wang, Y.; Zhou, B. Epithelial-mesenchymal transition in breast cancer progression and metastasis. Chin. J. Cancer 2011, 9, 603–611.
- Carver, E.A.; Jiang, R.L.; Lan, Y.; Oram, K.F.; Gridley, T. The mouse snail gene encodes a key regulator of the epithelial-mesenchymal transition. Mol. Cell. Biol. 2001, 21, 8184–8188.
- Barrallo-Gimeno, A.; Nieto, M.A. The Snail genes as inducers of cell movement and survival: Implications in development and cancer. Development 2005, 132, 3151–3161.
- Cano, A.; Perez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor Snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83.
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939.
- Kang, Y.B.; Massague, J. Epithelial-mesenchymal transitions: Twist in development and metastasis. Cell 2004, 118, 277–279.
- Peinado, H.; Olmeda, D.; Cano, A. Snail, ZEB and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428.
- Dave, N.; Guaita-Esteruelas, S.; Gutarra, S.; Frias, A.; Beltran, M.; Peiro, S.; de Herreros, A.G. Functional Cooperation between Snail1 and Twist in the Regulation of ZEB1 Expression during Epithelial to Mesenchymal Transition. J. Biol. Chem. 2011, 286, 12024–12032.
- Iorio, M.V.; Croce, C.M. MicroRNAs in Cancer: Small Molecules With a Huge Impact. J. Clin. Oncol. 2009, 27, 5848–5856.
- Dhamija, S.; Diederichs, S. From junk to master regulators of invasion: LncRNA functions in migration, EMT and metastasis. Int. J. Cancer 2016, 139, 269–280.
- Saxena, M.; Hisano, M.; Neutzner, M.; Diepenbruck, M.; Ivanek, R.; Sharma, K.; Kalathur, R.K.R.; Bürglin, T.R.; Risoli, S.; Christofori, G. The long non-coding RNA ET-20 mediates EMT by impairing desmosomes in breast cancer cells. J. Cell Sci. 2021, 134, jcs258418.
- Wang, Y.; Liu, J.; Huang, B.; Xu, Y.M.; Li, J.; Huang, L.F.; Lin, J.; Zhang, J.; Min, Q.H.; Yang, W.M.; et al. Mechanism of alternative splicing and its regulation. Biomed. Rep. 2015, 3, 152–158.
- Black, D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003, 72, 291–336.
- Lin, J.C.; Tsao, M.F.; Lin, Y.J. Differential Impacts of Alternative Splicing Networks on Apoptosis. Int. J. Mol. Sci. 2016, 17, 2097.
- Hertel, K.J. Combinatorial control of exon recognition. J. Biol. Chem. 2008, 283, 1211–1215.
- Lee, Y.; Rio, D.C. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu. Rev. Biochem. 2015, 84, 291–323.
- Liu, Z.J.; Yuan, G.X.; Liu, S.; Jia, J.T.; Cheng, L.Q.; Qi, D.M.; Shen, S.H.; Peng, X.J.; Liu, G.S. Identified of a novel cis-element regulating the alternative splicing of LcDREB2. Sci. Rep. 2017, 7, 46106.
- Wegener, M.; Müller-Mcnicoll, M. View from an mRNP: The Roles of SR Proteins in Assembly, Maturation and Turnover; Springer International Publishing: Berlin/Heidelberg, Germany, 2019; pp. 83–112.
- Marasco, L.E.; Kornblihtt, A.R. The physiology of alternative splicing. Nat. Rev. Mol. Cell Biol. 2023, 24, 242–254.
- Matsuda, Y.; Ueda, J.; Ishiwata, T. Fibroblast growth factor receptor 2: Expression, roles, and potential as a novel molecular target for colorectal cancer. Pathol. Res. Int. 2012, 2012, 574768.
- Eswarakumar, V.P.; Lax, I.; Schlessinger, J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149.
- Muh, S.J.; Hovhannisyan, R.H.; Carstens, R.P. A non-sequence-specific double-stranded RNA structural element regulates splicing of two mutually exclusive exons of fibroblast growth factor receptor 2 (FGFR2). J. Biol. Chem. 2002, 277, 50143–50154.
- Ranieri, D.; Rosato, B.; Nanni, M.; Magenta, A.; Belleudi, F.; Torrisi, M.R. Expression of the FGFR2 mesenchymal splicing variant in epithelial cells drives epithelial-mesenchymal transition. Oncotarget 2016, 7, 5440–5460.
- Katoh, M. FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review). Int. J. Mol. Med. 2016, 38, 3–15.
- Helsten, T.; Schwaederle, M.; Kurzrock, R. Fibroblast growth factor receptor signaling in hereditary and neoplastic disease: Biologic and clinical implications. Cancer Metastasis Rev. 2015, 34, 479–496.
- Katoh, Y.; Katoh, M. FGFR2-related pathogenesis and FGFR2-targeted therapeutics. Int. J. Mol. Med. 2009, 23, 307–311.
- Lu, C.M.; Huguley, S.; Cui, C.; Cabaniss, L.B.; Waite, P.D.; Sarver, D.M.; Mamaeva, O.A.; MacDougall, M. Effects of FGFR Signaling on Cell Proliferation and Differentiation of Apert Dental Cells. Cells Tissues Organs 2015, 201, 26–37.
- Tarkkonen, K.M.; Nilsson, E.M.; Kahkonen, T.E.; Dey, J.H.; Heikkila, J.E.; Tuomela, J.M.; Liu, Q.; Hynes, N.E.; Harkonen, P.L. Differential Roles of Fibroblast Growth Factor Receptors (FGFR) 1, 2 and 3 in the Regulation of S115 Breast Cancer Cell Growth. PLoS ONE 2012, 7, e49970.
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142.
- Warzecha, C.C.; Sato, T.K.; Nabet, B.; Hogenesch, J.B.; Carstens, R.P. ESRP1 and ESRP2 Are Epithelial Cell-Type-Specific Regulators of FGFR2 Splicing. Mol. Cell 2009, 33, 591–601.
- Ma, Q.; Zhang, K.; Guin, S.; Zhou, Y.-Q.; Wang, M.-H. Deletion or insertion in the first immunoglobulin-plexin-transcription (IPT) domain differentially regulates expression and tumorigenic activities of RON receptor Tyrosine Kinase. Mol. Cancer 2010, 9, 307.
- Ni, J.; Cozzi, P.J.; Hao, J.L.; Beretov, J.; Chang, L.; Duan, W.; Shigdar, S.; Delprado, W.J.; Graham, P.H.; Bucci, J.; et al. CD44 variant 6 is associated with prostate cancer metastasis and chemo-/radioresistance. Prostate 2014, 74, 602–617.
- Banky, B.; Raso-Barnett, L.; Barbai, T.; Timar, J.; Becsagh, P.; Raso, E. Characteristics of CD44 alternative splice pattern in the course of human colorectal adenocarcinoma progression. Mol. Cancer 2012, 11, 83.
- Ozawa, M.; Ichikawa, Y.; Zheng, Y.W.; Oshima, T.; Miyata, H.; Nakazawa, K.; Guan, H.B.; Shiozawa, M.; Akaike, M.; Watanabe, K.; et al. Prognostic significance of CD44 variant 2 upregulation in colorectal cancer. Br. J. Cancer 2014, 111, 365–374.
- Guo, W.; Frenette, P.S. Alternative CD44 splicing in intestinal stem cells and tumorigenesis. Oncogene 2014, 33, 537–538.
- Ishimoto, T.; Nagano, O.; Yae, T.; Tamada, M.; Motohara, T.; Oshima, H.; Oshima, M.; Ikeda, T.; Asaba, R.; Yagi, H.; et al. CD44 Variant Regulates Redox Status in Cancer Cells by Stabilizing the xCT Subunit of System xc(-) and Thereby Promotes Tumor Growth. Cancer Cell 2011, 19, 387–400.
- Zhang, H.H.; Brown, R.L.; Wei, Y.; Zhao, P.; Liu, S.L.; Liu, X.; Deng, Y.; Hu, X.H.; Zhang, J.; Gao, X.D.; et al. CD44 splice isoform switching determines breast cancer stem cell state. Genes Dev. 2019, 33, 166–179.
- Yanagisawa, M.; Huveldt, D.; Kreinest, P.; Lohse, C.M.; Cheville, J.C.; Parker, A.S.; Copland, J.A.; Anastasiadis, P.Z. A p120 catenin isoform switch affects Rho activity, induces tumor cell invasion, and predicts metastatic disease. J. Biol. Chem. 2008, 283, 18344–18354.
- Liu, D.S.; Zhang, H.; Cui, M.M.; Chen, C.S.; Feng, Y. Hsa-miR-425-5p promotes tumor growth and metastasis by activating the CTNND1-mediated beta-catenin pathway and EMT in colorectal cancer. Cell Cycle 2020, 19, 1917–1927.
- Keirsebilck, A.; Bonne, S.; Staes, K.; van Hengel, J.; Nollet, F.; Reynolds, A.; van Roy, F. Molecular cloning of the human p120(ctn) catenin gene (CTNND1): Expression of multiple alternatively spliced isoforms. Genomics 1998, 50, 129–146.
- Venhuizen, J.H.; Sommer, S.; Spann, P.N.; Friedl, P.; Zegers, M.M. Differential expression of p120-catenin 1 and 3 isoforms in epithelial tissues. Sci. Rep. 2019, 9, 90.
- Göttgens, E.L.; Span, P.N.; Zegers, M.M. Chapter Four—Roles and Regulation of Epithelial Splicing Regulatory Proteins 1 and 2 in Epithelial–Mesenchymal Transition. In International Review of Cell and Molecular Biology; Jeon, K.W., Galluzzi, L., Eds.; Academic Press: Cambridge, MA, USA, 2016; Volume 327, pp. 163–194.
- Paredes, J.; Correia, A.L.; Ribeiro, A.S.; Schmitt, F. Expression of p120-catenin isoforms correlates with genomic and transcriptional phenotype of breast cancer cell lines. Anal. Cell. Pathol. 2007, 29, 467–476.
- Du, J.P.; Zhu, Z.X.; Xu, L.; Chen, X.; Li, X.F.; Lan, T.; Li, W.; Yuan, K.F.; Zeng, Y. ARHGEF11 promotes proliferation and epithelial-mesenchymal transition of hepatocellular carcinoma through activation of beta-catenin pathway. Aging 2020, 12, 20235–20253.
- Wang, L.Z.; Habuchi, T.; Takahashi, T.; Mitsumori, K.; Kamoto, T.; Kakehi, Y.; Kakinuma, H.; Sato, K.; Nakamura, A.; Ogawa, O.; et al. Cyclin D1 gene polymorphism is associated with an increased risk of urinary bladder cancer. Carcinogenesis 2002, 23, 257–264.
- Paronetto, M.P.; Cappellari, M.; Busa, R.; Pedrotti, S.; Vitali, R.; Comstock, C.; Hyslop, T.; Knudsen, K.E.; Sette, C. Alternative Splicing of the Cyclin D1 Proto-Oncogene Is Regulated by the RNA-Binding Protein Sam68. Cancer Res. 2010, 70, 229–239.
- Kong, S.M.; Wei, Q.Y.; Amos, C.I.; Lynch, P.M.; Levin, B.; Zong, J.H.; Frazier, M.L. Cyclin D1 polymorphism and increased risk of colorectal cancer at young age. JNCI-J. Natl. Cancer Inst. 2001, 93, 1106–1108.
- Lu, F.M.; Gladden, A.B.; Diehl, J.A. An alternatively spliced cyclin D1 isoform, cyclin D1b, is a nuclear oncogene. Cancer Res. 2003, 63, 7056–7061.
- Urbanski, L.M.; Leclair, N.; Anczukow, O. Alternative-splicing defects in cancer: Splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics. Wiley Interdiscip. Rev.-RNA 2018, 9, e1476.
- Kong, D.J.; Sethi, S.; Li, Y.W.; Chen, W.; Sakr, W.A.; Heath, E.; Sarkar, F.H. Androgen Receptor Splice Variants Contribute to Prostate Cancer Aggressiveness Through Induction of EMT and Expression of Stem Cell Marker Genes. Prostate 2015, 75, 161–174.
- Qiu, Y.; Sun, F.; Yang, X. Androgen receptor splice variant AR3 promotes prostate cancer via modulating expression of autocrine/paracrine factors. Cancer Res. 2014, 74, LB-26.
- Guo, Z.Y.; Yang, X.; Sun, F.; Jiang, R.C.; Linn, D.E.; Chen, H.G.; Kong, X.T.; Melamed, J.; Tepper, C.G.; Kung, H.J.; et al. A Novel Androgen Receptor Splice Variant Is Up-regulated during Prostate Cancer Progression and Promotes Androgen Depletion-Resistant Growth. Cancer Res. 2009, 69, 2305–2313.
- Tabaglio, T.; Low, D.H.P.; Teo, W.K.L.; Goy, P.A.; Cywoniuk, P.; Wollmann, H.; Ho, J.; Tan, D.; Aw, J.; Pavesi, A.; et al. MBNL1 alternative splicing isoforms play opposing roles in cancer. Life Sci. Alliance 2018, 1, e201800157.
- Dittmar, K.A.; Jiang, P.; Park, J.W.; Amirikian, K.; Wan, J.; Shen, S.H.; Xing, Y.; Carstens, R.P. Genome-Wide Determination of a Broad ESRP-Regulated Posttranscriptional Network by High-Throughput Sequencing. Mol. Cell. Biol. 2012, 32, 1468–1482.
- Warzecha, C.C.; Carstens, R.P. Complex changes in alternative pre-mRNA splicing play a central role in the epithelial-to-mesenchymal transition (EMT). Semin. Cancer Biol. 2012, 22, 417–427.
- Braeutigam, C.; Rago, L.; Rolke, A.; Waldmeier, L.; Christofori, G.; Winter, J. The RNA-binding protein Rbfox2: An essential regulator of EMT-driven alternative splicing and a mediator of cellular invasion. Oncogene 2014, 33, 1082–1092.
- Shapiro, I.M.; Cheng, A.W.; Flytzanis, N.C.; Balsamo, M.; Condeelis, J.S.; Oktay, M.H.; Burge, C.B.; Gertler, F.B. An EMT-Driven Alternative Splicing Program Occurs in Human Breast Cancer and Modulates Cellular Phenotype. PLoS Genet. 2011, 7, e1002218.
- Venables, J.P.; Brosseau, J.P.; Gadea, G.; Klinck, R.; Prinos, P.; Beaulieu, J.F.; Lapointe, E.; Durand, M.; Thibault, P.; Tremblay, K.; et al. RBFOX2 Is an Important Regulator of Mesenchymal Tissue-Specific Splicing in both Normal and Cancer Tissues. Mol. Cell. Biol. 2013, 33, 396–405.
- Das, S.; Krainer, A.R. Emerging Functions of SRSF1, Splicing Factor and Oncoprotein, in RNA Metabolism and Cancer. Mol. Cancer Res. 2014, 12, 1195–1204.
- Martinez-Terroba, E.; Ezponda, T.; Bertolo, C.; Sainz, C.; Remirez, A.; Agorreta, J.; Garmendia, I.; Behrens, C.; Pio, R.; Wistuba, I.I.; et al. The oncogenic RNA-binding protein SRSF1 regulates LIG1 in non-small cell lung cancer. Lab. Investig. 2018, 98, 1562–1574.
- Tang, S.L.; Zhao, Y.R.; He, X.R.; Zhu, J.H.; Chen, S.; Wen, J.K.; Deng, Y.Q. Identification of NOVA family proteins as novel beta-catenin RNA-binding proteins that promote epithelial-mesenchymal transition. RNA Biol. 2020, 17, 881–891.
- Fish, L.; Pencheva, N.; Goodarzi, H.; Tran, H.; Yoshida, M.; Tavazoie, S.F. Muscleblind-like 1 suppresses breast cancer metastatic colonization and stabilizes metastasis suppressor transcripts. Genes Dev. 2016, 30, 386–398.