Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Gastroenterology & Hepatology
Metabolic-dysfunction-associated steatotic liver disease (MASLD) is a chronic liver disease that affects more than a quarter of the global population and whose prevalence is increasing worldwide due to the pandemic of obesity. Obesity, impaired glucose metabolism, high blood pressure and atherogenic dyslipidemia are risk factors for MASLD.
- cardiovascular disease
- fatty acids
- metabolic-dysfunction-associated steatotic liver disease
1. Introduction
The principal limitations of the terms nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH) are the reliance on exclusionary confounder terms and the use of potentially stigmatizing language. The name chosen to replace NAFLD was metabolic-dysfunction-associated steatotic liver disease (MASLD) [1]. There was consensus to change the definition to include the presence of steatotic liver disease and at least one of five cardiometabolic risk factors, which include (1) increase in body mass index (BMI) or waist circumference (WC); (2) impaired glucose metabolism; (3) high blood pressure; (4) high triglyceride (TG) levels; (5) low high-density cholesterol (HDL-C) levels [1]. The epidemiology and demographic characteristics of MASLD vary worldwide, usually parallel to the prevalence of obesity; however, a substantial proportion of patients are lean [2]. Individuals with MASLD have a high frequency of metabolic comorbidities and could place a growing strain on health-care systems all over the world. Prevalence data from 245 articles involving 2,699,627 persons were used with a hierarchical Bayesian approach to forecast the prevalence of MASLD through to 2040 [3]. By 2040, over half the adult population is forecasted to have MASLD [3]. The pandemic of obesity and its cardiometabolic consequences contribute to an increased prevalence of MASLD [4]. Approximately 20–30% of MASLD patients develop metabolic-dysfunction-associated steatohepatitis (MASH), leading to liver cirrhosis and associated complications, including hepatocellular carcinoma [5]. The worldwide disease burden from liver fibrosis due to MASLD is expected to increase around two to three-fold within a decade.
Furthermore, MASLD patients have cardiometabolic risk factors and are predisposed to liver fibrosis as well as atherosclerotic cardiovascular disease (ASCVD). Yoneda, M. et al. retrospectively analyzed data for 2,452,949 people to estimate the relationship between CVD and MASLD [6]. The incidence rates of CVD were 1.01 (95%CI (confidence interval), 0.98 to 1.03) and 2.69 (95%CI, 2.55 to 2.83) per 1000 person-years in the non-MASLD and MASLD groups, respectively. The overall prevalence of hypertriglyceridemia and diabetes was 13.6 and 4.3%, respectively, in the non-MASLD group and 64.1 and 20.6%, respectively, in the MASLD group. The CVD risk increased with hypertriglyceridemia and diabetes.
2. The Metabolic Abnormalities That Induce MASLD
The features of metabolic syndrome are not only highly prevalent in patients with MASLD but components of metabolic syndrome also increase the risk of developing MASLD [5]. The established conditions for developing MASLD include obesity, type 2 diabetes, hypertension and dyslipidemia such as high TG and low HDL-C levels [5].
Obesity is the most common and well-documented risk factor for MASLD. To investigate whether central obesity is associated with MASLD formation after adjusting for general obesity, a meta-analysis was performed [7]. In the meta-analysis, which used twenty eligible studies, the pooled odds ratio (OR) in WC and BMI were 2.34 (95%CI, 1.83 to 3.00) and 2.85 (95%CI, 1.60 to 5.08), respectively [7].
Although MASDL, MASH and MASH with advanced fibrosis are closely associated with type 2 diabetes, their global prevalence rates have not been well described. To estimate the prevalence of MASLD, MASH and advanced fibrosis among patients with type 2 diabetes, a meta-analysis using 80 studies from 20 countries was performed [8]. The global prevalence of MASLD among patients with type 2 diabetes was 55.5% (95%CI, 47.3 to 63.7) and the global prevalence of MASH among individuals with type 2 diabetes was 37.3% (95%CI, 24.7 to 50.0) [8].
An increasing body of evidence connects MASLD to hypertension. To estimate the nature and magnitude of the association between MASLD and hypertension, a meta-analysis using 11 studies was performed [9]. The presence of hypertension was significantly associated with an increased risk of incident MASLD (hazard ratio (HR), 1.63; 95%CI, 1.41 to 1.88) [9]. Pooled analysis showed that the presence of MASLD was significantly associated with an increased incidence of hypertension (HR, 1.55; 95%CI, 1.29 to 1.87) [9], indicating the existence of a bidirectional relationship between MASLD and hypertension that was independent of traditional cardiometabolic risk factors.
Dyslipidemia, which includes high serum TG levels and low serum HDL-C levels, is also common in patients with MASLD. The prevalence of MASLD in individuals with dyslipidemia attending lipid clinics has been estimated to be 50% [10].
Metabolic syndrome and its components such as obesity, impaired glucose metabolism, high blood pressure and dyslipidemia are all closely associated with insulin resistance. The pathogenesis of MASLD largely remains unknown. Sanyal, A.J. et al. demonstrated that peripheral insulin resistance was present in both MASLD and MASH patients by using a hyper-insulinemic euglycemic clamp [11]. Many investigations have shown that defects in the insulin signaling pathway, especially those associated with insulin receptor substrate-2 (IRS-2), are definitely implicated in the pathogenesis of insulin resistance [12]. Rats with MASLD developed insulin resistance, showing increased fasting blood glucose and insulin levels, increased weight of epididymal fat, obvious hepatic steatosis and inflammation and down-regulated IRS-2 mRNA and protein levels compared with normal controls [13]. Further, an insulin sensitizer, pioglitazone, underwent significant recovery, including up-regulated IRS-2 mRNA and protein levels [13]. Insulin resistance may greatly contribute to the development of MASLD.
The effects of lipid metabolism abnormalities induced by insulin resistance on the development of MASLD are shown in Figure 1. Accumulated visceral adipose tissue produces more inflammatory cytokines, such as tumor necrosis factor alpha (TNF-a), interleukin-6 (IL-6) and IL-1b, and less adiponectin, which induces systemic insulin resistance [14]. The metabolism of free fatty acids (FFAs) is altered in insulin resistance [15]. The enzymes lipoprotein lipase (LPL) and hormone-sensitive lipase (HSL) are rate-limiting factors for TG and FA metabolism because LPL hydrolyzes extracellular TG in lipoproteins and HSL hydrolyzes intracellular TG in adipocytes [16].
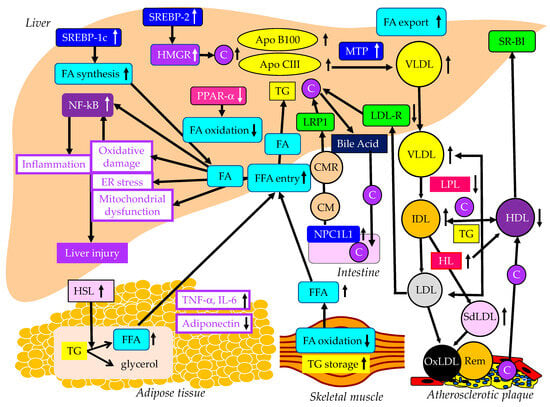
Figure 1. The abnormal lipid metabolism possibly induced by insulin resistance and its association with the development of MASLD. Black and white arrows pointing upward and downward indicate an increase or decrease in expression or activity, respectively. Solid black lines indicate the flow of substances and the effects of each metabolic event.
Insulin resistance enhances the expression and activity of HSL in adipose tissue. HSL catalyzes the hydrolysis of TG into FFA [17]. Insulin resistance is closely associated with an excess TG storage within the skeletal muscle [16]. Insulin resistance reduces FA oxidation, leading to diminished use of FAs and storage of TG within the skeletal muscle. Serum FFAs increase due to increased release from the adipose tissue and decreased FA use in the skeletal muscle. An increased amount of FFA enters the liver, leading to overproduction of TG-rich lipoproteins such as very-low-density lipoprotein (VLDL). Insulin resistance is associated with reduced apo B100 degradation [18] and elevated hepatic apo CIII production [19], which increase VLDL because both apo B100 and apo CIII constitute VLDL. Insulin resistance increases the expression of microsomal TG transfer protein (MTP), a key enzyme involved in VLDL assembly [18]. In an insulin-resistant state, an increased FFA entry to liver, reduced degradation of apo B100 and enhanced expression of apo CIII and MTP may elevate hepatic production of VLDL. Insulin resistance also causes an increased expression of sterol regulatory element binding protein 1c (SREBP-1c), which increases FA synthesis [20]. Hepatic FA metabolism is regulated by a combination of FA uptake, FA export by VLDL secretion, de novo FA synthesis by SREBP-1c and FA utilization by β-oxidation. FA accumulation is one of the features of MASLD.
Two major physically distinct species of VLDL exist: larger TG-rich VLDL1 and smaller VLDL2 [21]. At normal TG concentrations, VLDL1 and VLDL2 circulate in approximately equal proportions. Hepatic TG accumulation and insulin resistance increase VLDL1 secretion [22,23]. MASH patients have been shown to have more pronounced postprandial intestinal and hepatic VLDL1 accumulation, LDL lipid peroxidation and reduced total antioxidant status [24]. Postprandial intestinal VLDL1 independently predicted oxidized LDL (OxLDL) and reduced total antioxidant status responses in MASH. Postprandial intestinal VLDL1 accumulation is associated with a pro-oxidant imbalance in MASH, and both correlate with the severity of liver disease. Otsuka Long-Evans Tokushima fatty rats showed overproduction of VLDL compared with control rats [25]. In the livers of these rats, the mRNA levels of TNF-α, IL-1b and IL-6 were increased and the mRNA, protein, and tyrosine phosphorylation levels of IRS-2 were decreased. Overproduction of VLDL in the liver is significantly associated with hepatic oxidative stress, inflammation and insulin resistance. However, it remains unclear whether VLDL itself has the property of enhancing such exacerbating factors of liver fibrosis or whether metabolic abnormalities that induce VLDL overproduction promote liver fibrosis. TG accumulation and VLDL overproduction are also features of MASLD.
MTP is predominantly expressed in hepatocytes and enterocytes and is required for the assembly and secretion of VLDL. A rare causal variant in the MTP gene that was associated with progressive MASLD, unrelated to metabolic syndrome, was identified [26]. Hepatocyte-like cells derived from a homozygote donor had significantly lower MTP activity and lower lipoprotein apo B secretion than wild-type cells. Cytoplasmic TG accumulation in hepatocyte-like cells triggered endoplasmic reticulum stress, secretion of pro-inflammatory mediators and production of reactive oxygen species (ROS). This MTP gene variant was associated with progressive MASLD. Increased expression of MTP can be beneficial for protection against MASLD. Cytoplasmic TG accumulation may induce MASLD.
FA oxidation primarily occurs in the mitochondria; however, FA oxidation commences in the peroxisomes and then is finally processed in the mitochondria [27]. In obesity, ω-oxidation by cytochrome P450 enzymes also contributes to FA oxidation. This pathway for FA oxidation generates large amounts of ROS [28]. The entry of FAs into mitochondria depends on carnitine palmitoyl-transferase 1 (CPT-1). One of the major regulators of CPT-1 is the peroxisome proliferator-activated receptor (PPAR)-α [29,30,31,32]. Activation of PPARα induces the transcription of genes related to FA oxidation [29,33,34]. Visceral adiposity and insulin resistance are negatively correlated with liver PPARα gene expression [34].
Overexpression of apo CIII, independent of a high-fat diet, produces MASLD-like features, including increased liver lipid content, decreased antioxidant capacity, increased expression of TNFα and IL-1β and decreased expression of adiponectin receptor [35]. A high-fat diet induced hepatic insulin resistance and marked increases in plasma TNFα (eight-fold) and IL-6 (60%) in apo-CIII-overexpressing mice [35]. Cell death and apoptosis were augmented in apo-CIII-overexpressing mice regardless of diet [35]. Fenofibrate treatment reversed several of the effects associated with diet and apo CIII expression but did not normalize inflammatory traits even when the liver lipid content was fully corrected [35]. An increase in apo CIII plays a major role in liver inflammation and cell death in MASLD. There are no reports on adverse effects for apo CIII deficiency in MASLD, and increased apo CIII may adversely affect MASLD.
An increase in FFAs leads to hepatic insulin resistance by interacting with insulin signaling [36,37]. The anti-lipolytic function of insulin is impaired in insulin resistance, which may facilitate hepatic TG synthesis. Saturated FAs generate lipotoxic intermediate products, such as diacylglycerols [38]. Lipotoxic intermediate products cause endoplasmic reticulum stress and ROS formation, which are major factors for the pathogenesis of MASH [39,40]. By binding to Toll-like receptor 4, saturated FAs induce the augmentation of mitochondrial dysfunction and activation of pro-inflammatory nuclear factor-kappa B (NF-κB) [39].
The studies using animal models, particularly those using molecular inhibition of TG synthesis [41], and the available small human lipidomic studies, have ruled out TG as the major lipotoxic mediator of MASH [42]. The focus now falls on other lipid species, particularly FFAs, diacylglycerol, toxic phospholipids (ceramides, sphingolipids) [42] and, most recently, cholesterol. Insulin resistance activates SREBP-2, which induces the expression of 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGR), the rate-limiting enzyme of cholesterol biosynthesis, resulting in increases in free cholesterol and cholesterol ester in the liver [43]. Such increased free cholesterol and cholesterol ester may induce inflammation and cell death [43].
3. The Association of MASLD with ASCVD
A retrospective analysis of 619 patients diagnosed with MASLD showed that CV events (38.3%), followed by non-liver malignancy (18.7%) and complications of liver cirrhosis (7.8%), were the three most common causes of death in MASLD patients [44], suggesting that CV events were the most crucial determinant of mortality in MASLD patients. A meta-analysis showed that MASLD was significantly associated with an increase in the development of CVD (odds ratio (OR), 2.05; 95% confidence interval (95%CI), 1.81 to 2.31; p < 0.0001) [45]. However, MASH has a higher liver-related (OR for MASH, 5.71; 95%CI, 2.31 to 14.13; OR for MASH with advanced fibrosis, 10.06; 95%CI, 4.35 to 23.25) but not cardiovascular mortality (OR, 0.91; 95%CI, 0.42 to 1.98). Therefore, patients with MASLD can be said to be a high-risk group for CVD as well as a high-risk group for developing MASH.
A multicenter large retrospective study showed that the BMIs of subjects with MASLD were significantly higher than in those without MASLD (p < 0.01) [46]. The prevalence of MASLD showed a linear increase corresponding to an increase in BMI (BMI < 23 kg/m2, 10.5%; BMI ≥ 23 kg/m2 and <25 kg/m2, 37.9%; BMI ≥ 25 kg/m2 and <28 kg/m2, 58.4%; BMI ≥ 28 kg/m2, 84.2%) [46]. In short, a 7.4–11.4% increase in the prevalence of MASLD per 1 kg/m2 of BMI was observed. The prevalence of MASLD showed a linear increase when serum TG and LDL-C levels were increased and a linear decrease when HDL-C levels were increased. The prevalence of MASLD was 22.8% in subjects with normal TG levels (<150 mg/dL) and 59.5% in subjects with hypertriglyceridemia (>150 mg/dL). The prevalence of MASLD was 27.3% in subjects with normal HDL-C levels (>40 mg/dL) and 61.7% in subjects with hypo-HDL-C (<40 mg/dL). The prevalence of MASLD was 26.4% in subjects with normal LDL-C (<140 mg/dL) and 38.5% in subjects with hyper-LDL-C (>140 mg/dL) [46].
The increased production of VLDL observed in MASLD is caused by insulin resistance as described above, and insulin resistance reduces the degradation of VLDL in the blood (Figure 1). Insulin resistance adversely affects enzymes such as LPL and hepatic lipase (HL), leading to conditions that are highly atherogenic, such as a decrease in HDL and increases in small-dense LDL (SdLDL) and remnant lipoproteins [47]. Insulin resistance reduces LPL activity [47]. LPL is the rate-limiting enzyme for the catabolism of TG-rich lipoproteins such as VLDL and intermediate-density lipoprotein (IDL) [48]. The formation of HDL is related to the catabolism of TG-rich lipoproteins by LPL [49]. Therefore, reduced LPL activity increases VLDL and IDL and reduces HDL. The activity of HL, the enzyme that facilitates the catabolism of HDL, is correlated with insulin resistance [50]. Low serum HDL-C levels may be partially due to an increased clearance by HL [50]. LDL size is inversely proportional to HL activity [51], and patients with high HL have more SdLDL as compared with subjects with low HL activity [52]. Increased HL activity due to insulin resistance may increase atherogenic lipoprotein and SdLDL. Remnant lipoproteins have undergone extensive intravascular remodeling. LPL, HL and cholesterol ester transfer protein (CETP) induce structural and atherogenic changes that distinguish remnant lipoproteins from non-remnant lipoproteins [21]. Via the LPL-mediated removal of TG and the CETP-mediated exchange of TG for cholesterol from LDL and HDL, remnant lipoproteins contain more cholesterol than nascent VLDL [53].
HDL plays a role in reverse cholesterol transport from atherosclerotic plaques, which is an anti-atherogenic effect [54]. Therefore, reduced HDL induces an atherogenic status. Since SdLDL is not recognized by the LDL receptor, SdLDL stays in blood for a longer period [54]. SdLDL is likely to be adhesive to the endothelium and migrate into the subendothelial space. SdLDL is easily oxidized because of a lack of antioxidative capacity [54]. LDL and SdLDL are taken up by macrophages via a scavenger receptor after oxidative modification [54]. Remnant lipoproteins are taken up by macrophages without modification such as oxidation, which is a highly atherogenic property.
Insulin modulates LDL receptor expression and activity. The inactivity of insulin represses LDL receptor transcription [55], which can explain the increase in LDL-C in the insulin-resistant state. Niemann-Pick C1-like 1 (NPC1L1) plays a pivotal role in intestinal cholesterol absorption. The expression of NPC1L1 was investigated in non-diabetic rats and diabetic cholesterol-fed rats [56]. There was a positive correlation between intestinal NPC1L1 mRNA and chylomicron (CM) cholesterol. LDL receptor-related protein 1 (LRP1) is an endocytic and signaling receptor expressed in several tissues that plays a crucial role in clearance of CM remnants from circulation [57]. Furthermore, LDL and other cholesterol-rich, apo B-containing lipoproteins, once they become retained and modified within the arterial wall, cause atherosclerosis [58].
This entry is adapted from the peer-reviewed paper 10.3390/ijms242015473
This entry is offline, you can click here to edit this entry!