Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
Human epidermal growth factor receptor 3 (HER3) is the only family member of the EGRF/HER family of receptor tyrosine kinases that lacks an active kinase domain (KD), which makes it an obligate binding partner with other receptors for its oncogenic role. When HER3 is activated in a ligand-dependent (NRG1/HRG) or independent manner, it can bind to other receptors (the most potent binding partner is HER2) to regulate many biological functions (growth, survival, nutrient sensing, metabolic regulation, etc.) through the PI3K–AKT–mTOR pathway.
- breast cancer
- human epidermal growth factor receptor 3
1. Introduction
Although after 2020, the diagnosis and treatment of cancer were adversely affected due to the coronavirus disease 2019 (COVID-19) pandemic, in 2022, 1,918,030 new cancer cases and 609,360 cancer deaths are projected in the United States [1]. Despite the enormous efforts in advancing anti-cancer agents, it is still challenging to treat cancer due to the development of drug resistance. Accumulation of mutations in solid tumors imposes even more pharmacological challenges. The ability of tumor cells to survive, grow, migrate, and invade depends on the interaction of cell surface receptors and many growth factors [2]. Some of these growth factors were found to bind with cell surface localized receptor tyrosine kinases (RTKs), as shown in Figure 1. RTKs have been an attractive target for developing anti-cancer agents due to their accessibility and the ability to block the catalytic kinase function using small molecules [3,4,5]. There are four types of RTKs: human epidermal growth factor receptor (HER) 1 (EGFR, ErbB1), HER2 (Neu, ErbB2), HER3 (ErbB3), and HER4 (ErbB4). These RTKs are generally expressed in epithelial, mesenchymal, and neuronal tissues and are found to regulate cell division, proliferation, and differentiation [6,7]. Among these four members of RTKs, EGFR and HER2 are the most studied targeted molecules in cancer therapy [8].
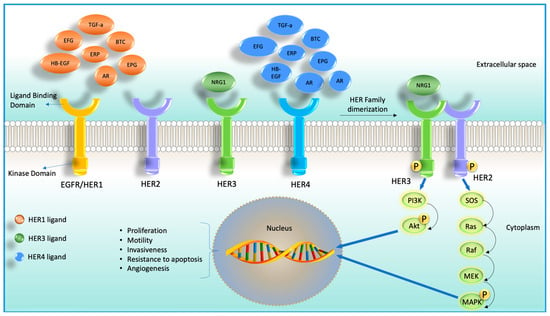
Figure 1. Cartoon diagram showing four receptor tyrosine kinases (RTKs) members and their corresponding ligands (HER2 does not have any ligand) on the left-hand side. On the right-hand side, it shows ligand-dependent heterodimerization of HER2 and HER3 and their downstream signaling cascade.
Although HER3 has long been underestimated for targeting, recent studies found its emerging role in oncogenesis, tumor progression, and drug resistance [9]. HER3 is a unique family member of HER family proteins for many reasons. Unlike other HER family members, HER3 cannot form a homodimer, lacks/almost no intracellular kinase activity, and can form a heterodimer with other non-HER family proteins [10,11,12,13]. When HER3 is activated via binding with other receptors, it primarily activates PI3K/Akt signaling [14,15]. Additionally, HER3 was also reported to activate the MAPK cascade, Janus kinase (JAK), and proto-oncogene c-Src (SRC) signaling pathways, all of these pathways are tumorigenic [16,17]. It is now well recognized that HER3 can restore the signaling function via the PI3K/Akt axis under EGFR-targeted inhibition [18].
Even though it is now recognized that HER3 is a prime target for therapy, all the drugs available in the market are targeted against only two members of the HER/ERBB family proteins, namely EGFR and HER2. As it is impossible to target HER3 via conventional methods through inhibition of kinase activity, many pharmaceutical companies have been trying to develop different agents, including monoclonal antibodies and drug conjugates. Nonetheless, many drugs are developed targeting HER3 and are currently in various stages of clinical development. Interestingly, some studies also suggest that the extracellular domain of HER3 is not required for signaling, which raises an important question about the applicability of that specific therapy that targets the extracellular domain of HER3 [19].
It is suggested that signal generation in the HER3 occurs through an asymmetric kinase dimerization, where one kinase allosterically activates the other kinase [20]. Although it has been assumed that signaling and oncogenic functions of HER3 involve heterodimerization with another receptor, no crystal structure has been developed for a fully active HER3 signaling complex, so it is still known whether HER3 forms a dimer or there are more than two binding partners involved for this interaction. In fact, higher-order oligomerization is the widely believed mode of signaling in the HER family. The involvement of oligomeric assemblies in HER family signaling has been suggested by several studies involving fluorescent tracing and other techniques [21,22,23,24,25,26,27]. However, the interfaces involved in these interactions and their functional relevance remain to be defined. Hence, it is essential to critically review the 30 years of bench-to-bedside research to develop novel pharmacological interceptors of HER3. Such understanding can lay the foundation for a newer generation of drugs that can significantly enhance the efficacy of TKIs and prove highly effective without the need for cytotoxic agents.
2. HER3 Structure and Mechanism of Activation
HER3 was discovered by Kraus et al. in 1989, and it maps to chromosome 12q13 [28]. HER3 protein mainly has five domains: the extracellular domain, transmembrane domain, juxta membrane domain, kinase domain, and C-terminal tail, as shown in Figure 2. The extracellular domain consists of four subdomains, from domain I to domain IV. Upon ligand binding, a structural conformational change occurs that converts an untethered/inactive form to a tethered/active form, which enables it for heterodimerization with other HER receptors. Although the HER3 kinase domain (KD) shares 83% amino acid sequence identity with both EGFR and HER2, the presence of specific nonconservative amino acid residues (Cys-721, His-740, and Asn-815) in the HER3 KD makes its KD inactive [29].
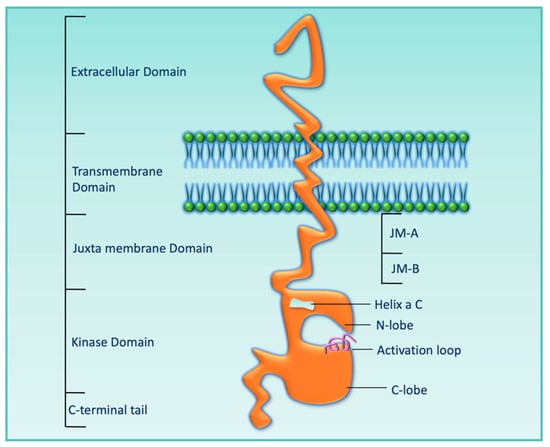
Figure 2. Cartoon diagram showing the extracellular domain, transmembrane domain, juxta membrane domain, kinase domain, and C-terminal tail of HER3.
Similar to other receptor tyrosine kinases (RTKs), the extracellular domain of HER3 exists in a reversible equilibrium between open (active) and closed (inactive) conformations. HER3 is active when this equilibrium shifts towards open conformation, allowing its dimerization arm within domain II to be exposed for dimerization (through the cysteine-rich CR1 region). So, when HER3 is in the open conform, it can dimerize with another HER family receptor. HER3 is the only family member of the HER family that does not form a homodimer. HER3 activation can occur in a ligand-dependent or ligand-independent manner. Conventionally, HER3 becomes activated via binding of its ligand NRG1 (also known as HRG) to the extracellular domain of HER3, which exposes its dimerization interface to be available for binding with other receptors. However, when any of its dimerization partners are present at sufficient concentration, it stabilizes HER3 in the open conformation transiently, known as ligand-independent activation.
Signal generation in the HER family occurs through an asymmetric kinase dimerization, where one kinase allosterically activates the other kinase. In the case of the HER2-HER3 dimer, HER2 acts as a receiver kinase, phosphorylating tyrosine residues on the C-terminal tails of the activator kinases, i.e., HER3. After ligand binding, HER3 heterodimerizes with other HER family proteins that cross the phosphorylate C-terminal tail of HER3. This transphosphorylation at the HER3 C-terminal tail creates docking sites for p85, a regulatory subunit of protein phosphatidylinositol 3-kinase (PI3K) [30]. Studies reported that six tyrosine residues of the HER3 C-terminal tail are phosphorylated to recruit and activate PI3K [15,31]. Once activated, PI3K phosphorylates phosphatidylinositol bisphosphate (PIP2) and is converted to phosphatidylinositol trisphosphate (PIP3) in the plasma membrane [32]. PKB/Akt binds to PIP3, allowing PDK1 to phosphorylate the T308 residue of Akt to activate it [32]. The PI3K/Akt pathway also activates the mammalian target of rapamycin (mTOR) to control various biological processes, including survival, translation, nutrient sensing, cell cycle control, and metabolic regulation [32].
3. Expressions of HER3 in Different Types of Cancers
HER3 expression and activation levels go up during organogenesis in the postnatal maturation period [33,34]. Additionally, embryonic HER3 knockdown mice showed severely underdeveloped sympathetic ganglia and a partial lack of Schwann cells [35]; these suggest the importance of HER3 in the development of the fetal mouse brain. HER3 transcripts were reported in the human liver, kidney, and brain but not in heart or lung fibroblasts [28]. Like EGFR and HER2 expressions, HER3 expression was also reported in normal keratinocyte and glandular epithelium tissues; however, unlike EGFR and HER2, HER3 expression was not detected in fibroblasts, skeletal muscle, or lymphoid cells [28], suggesting the tissue-specific function of HER3 in ectodermal development.
As physiological HER3 expression was reported in normal tissue, similarly, abnormal expression of HER3 has often been linked to a variety of different cancers, including breast cancer, colorectal carcinoma, squamous cell carcinoma of the head and neck, uveal melanoma, and gastric, ovarian, prostate, and bladder cancers, as shown in Table 1 [36,37,38,39]. Overexpression of HER3 protein has been linked to 50–70% of cases of breast cancer [40,41,42]. Upregulation of HER3 has been associated with metastasis, tumor volume, and risk of recurrence [43,44]. In colon cancer, HER3 overexpression has been associated with lymph node metastasis and poor progression [45,46,47,48]. Similarly, HER3 expression has been associated with increased metastasis and decreased overall survival in squamous cell carcinoma of the head and neck [49,50]. The above reports suggest the importance of HER3 in tumorigenesis and the need for targeting to inhibit tumor growth.
Although HER3 is a membrane protein (like EGFR), it has also been reported to localize in the nucleus. A study reported in human breast cancer cells that HER3 predominantly localizes in the nucleus; however, upon ligand stimulation, it is transported to the cytoplasm [51]. Previous studies suggested that HER3 localization also has tissue-specific functions. In gastric carcinomas, HER3 nuclear expression is associated with vascular and lymphatic invasion [52]. Similarly, in prostate cancer, HER3 nuclear expression is correlated with tumor progression [53]. In contrast, nuclear HER3 expression is associated with favorable overall survival in uveal melanoma [54]. In contrast, membranous expression of HER3 is associated with decreased survival in head and neck squamous cell carcinoma [55].
Table 1. Expression of HER3 in different types of cancer detected by immunohistochemistry (IHC) in the selected studies.
| Cancer Type | % of HER3 Overexpression | Antibody Used in IHC | Cutoff for Overexpression | Reference |
|---|---|---|---|---|
| Pancreatic | 41.3% | Nanotools, Teningen, Germany | Moderate staining is observed in >10% of tumor cells (score 2+), and strong staining is observed in >10% of tumor cells (score 3) | [56] |
| Breast | 43.0% | Clone 2F12, Labvision, Cheshire, UK | Positive: Optimal cutoffs for HER2:HER3 dimers were assessed by performing a minimum P value estimation using approximate 5% cutoffs across the entire dataset using relapse-free survival as an endpoint | [57] |
| 17.5% | IgG1, Neomarkers, UK | 4-point scale, where 0 = no staining, 1 = light staining, 2 = moderate staining, and 3 = strong staining | [58] | |
| Colorectal | 17.0% | MAb-MS-725-P, Neomarkers, Fremont, CA | Membranous staining: >1% of tumor cells stained. Cytoplasmic staining: 2+: moderate immunostaining in >10% of tumor cells and 3+: strong immunostaining in >10% of tumor cells | [59] |
| 69.7% | Lab Vision, Fremont, CA Cytoplasmic and membrane Cytoplasmic | Cytoplasmic staining: 0: no staining or weak staining in <10% of tumor cells. membranous staining: 0: no staining in <10% of tumor cells; 1: weak staining in >10% tumor cells; 2+: moderate staining in > 10% tumor cells; 3: strong staining in >10% tumor cells |
[59] | |
| 20.9% | Clone C-17, 1:50; Santa Cruz, CA | Depending on the intensity of staining, HER3 expression was classified as weak, intermediate, or strong | [60] | |
| Gastric | 59.0% | Mouse monoclonal antibody, Neomarkers | 2+ = moderate staining, and 3+ = strong staining | [61] |
| 34.0% | RB-9211 rabbit polyclonal, dilution 1:100, N terminal; Neomarkers, Fremont, CA | 0 = <10% of positively stained cells; 1 = 10–25%; 2 = 26–50%; 3 = 51–75%; 4 = >75% | [52] | |
| Melanoma | approximately 65.0% | Clone C-17, 1:50 dilution; SantaCruz, CA | Positive: high GIS > 6 (GIS: German immunohistochemical scoring) | [60] |
| Ovary | 53.4% | C-17, rabbit polyclonal antibody; dilution: 1:25; Santa Cruz, CA | Positive: scores >8 | [62] |
| Head and Neck | 8.8% | RTJ.2, mouse monoclonal antibody; Santa Cruz, CA | Scores of 0, +1, +2, and +3 for increasing intensity | [55] |
| Cervix | 55.5% | MS-725-P, mouse antibody; Neomarkers | Intensity scale 0–4 based on pixel density | [63] |
4. Role of HER3 in the Genesis and Progression of Different Types of Cancer
As the KD of HER3 is inactive, it is an obligate binding partner with multiple HER family proteins to activate PI3K/Akt signaling [6]. PI3K/Akt signaling contributes to many biological functions (translation, survival, nutrient sensing, metabolic regulation, and cell cycle control) [64,65,66], which are also involved in tumorigenesis, suggesting the importance of HER3 in cancer development. This HER3/PI3 K/Akt signaling cascade has been linked to breast, ovarian, colon, gastric, and lung cancer [67].
Interestingly, a sequence analysis suggested the binding activity of HER3 cytoplasmic domains with not only PI3K but also other proteins such as GRB7, GRB2, SHC, and SRC [15]. GRB7 and GRB2 interact with HER3 through their SH2 domain, whereas SRC SHC interacts with HER3 through the PTB domain [67,68,69,70]. Studies found that GRB2 binds to HER3 only in the absence of GRB7 [69]. The SHC–HER3 interaction is essential for MAPK signaling [70].
Because HER3 function depends on binding with other receptors, HER3 cannot transform a normal cell into cancer cells [71,72]. The most favorable binding partner of HER3 is HER2. A study showed that introducing HER3 into NIH3T3 fibroblast cells results in a low level of colony growth, but when HER3 is transfected with HER2, it significantly induces colony growth compared to HER2 and HER3 alone [73]. Similarly, in vivo studies showed that the introduction of both EGFR and HER3 was not tumorigenic, whereas the introduction of HER2 and HER3 yielded significant tumorigenic growth compared to the combination of other HER family proteins. These suggest that the most potent binding partner of HER3 is HER2.
HER3 is similarly important to its most potent binding partner, HER2 [38]. Studies also found that the knockdown of HER3 expression is more effective in inhibiting breast cancer cell growth than the knockdown of EGFR [38,74]. Interestingly, HER3 was found to preferentially dimerize with EGFR to induce cell proliferation, invasion, and migration in melanoma and pancreatic cancer [60,75]. Activation of HER3 via neuregulin-1 (NRG-1) was found to be enriched in a subset of SCCHN, and HER3 expression was associated with reduced survival in SCCHN [55,76]. Moreover, our previous studies in a panel of cancer cell lines of breast, bladder, colon, gastric, esophageal, lung, tongue, and endometrium cancer showed constitutive phosphorylation of HER3 in most of the HER2-amplified cancers [77]. Also, we noticed upon HER3 knockdown that overall tumor growth was substantially reduced in three HER2-amplified cancers from non-breast origin, suggesting HER3 has a vital role in tumorigenesis and tumor growth beyond breast cancer [77].
This entry is adapted from the peer-reviewed paper 10.3390/cells12212517
This entry is offline, you can click here to edit this entry!