Schizophrenia is one of the most serious psychiatric disorders and is characterized by reductions in both brain volume and spine density in the frontal cortex. RhoA belongs to the RAS homolog (Rho) family and plays critical roles in neuronal development and structural plasticity via Rho-kinase. RhoA activity is regulated by GTPase-activating proteins (GAPs) and guanine nucleotide exchange factors (GEFs). Several variants in GAPs and GEFs associated with RhoA have been reported to be significantly associated with schizophrenia. Here, we summarize clinical evidence showing that variants in genes regulating RhoA activity are associated with schizophrenia.
- copy number variants
- CNVs
- single-nucleotide polymorphisms
1. Introduction
2. Rho Family Activity Is Regulated by GTPase-Activating Proteins (GAPs) and Guanine Nucleotide Exchange Factors (GEFs)
3. Rho Family Protein Effectors and Their Physiological Roles in the Brain
4. Schizophrenia-Associated Genes Involved in Small GTPase RhoA Signaling
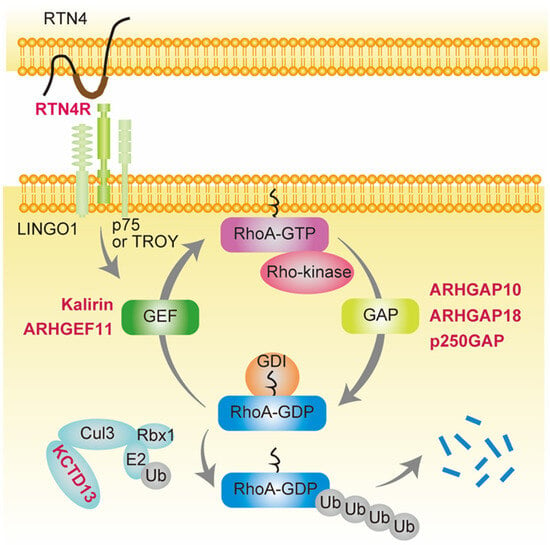
4.1. GAPs
4.1.1. ARHGAP10
4.1.2. ARHGAP18
4.1.3. p250GAP (ARHGAP32)
4.2. GEFs
4.2.1. KALRN
4.2.2. ARHGEF11
4.3. Others
4.3.1. RTN4R
4.3.2. 16p11.2 CNVs and the KCTD13-Cul3-RhoA Pathway
This entry is adapted from the peer-reviewed paper 10.3390/ijms242115623
References
- Jauhar, S.; Johnstone, M.; McKenna, P.J. Schizophrenia. Lancet 2022, 399, 473–486.
- Gogtay, N.; Vyas, N.S.; Testa, R.; Wood, S.J.; Pantelis, C. Age of onset of schizophrenia: Perspectives from structural neuroimaging studies. Schizophr. Bull. 2011, 37, 504–513.
- Stępnicki, P.; Kondej, M.; Kaczor, A.A. Current Concepts and Treatments of Schizophrenia. Molecules 2018, 23, 2087.
- Haijma, S.V.; Van Haren, N.; Cahn, W.; Koolschijn, P.C.M.P.; Pol, H.E.H.; Kahn, R.S. Brain volumes in schizophrenia: A meta-analysis in over 18,000 subjects. Schizophr. Bull. 2012, 39, 1129–1138.
- Howes, O.D.; Cummings, C.; Chapman, G.E.; Shatalina, E. Neuroimaging in schizophrenia: An overview of findings and their implications for synaptic changes. Neuropsychopharmacology 2022, 48, 151–167.
- Glausier, J.; Lewis, D. Dendritic spine pathology in schizophrenia. Neuroscience 2013, 251, 90–107.
- Broadbelt, K.; Byne, W.; Jones, L.B. Evidence for a decrease in basilar dendrites of pyramidal cells in schizophrenic medial prefrontal cortex. Schizophr. Res. 2002, 58, 75–81.
- Konopaske, G.T.; Lange, N.; Coyle, J.T.; Benes, F.M. Prefrontal cortical dendritic spine pathology in schizophrenia and bipolar disorder. JAMA Psychiatry 2014, 71, 1323–1331.
- Runge, K.; Cardoso, C.; de Chevigny, A. Dendritic Spine Plasticity: Function and Mechanisms. Front. Synaptic Neurosci. 2020, 12, 36.
- Weinberger, D.R.; Berman, K.F.; Zec, R.F. Physiologic dysfunction of dorsolateral prefrontal cortex in schizophrenia: I. Regional cerebral blood flow evidence. Arch. Gen. Psychiatry 1986, 43, 114–124.
- Haddad, P.M.; Correll, C.U. The acute efficacy of antipsychotics in schizophrenia: A review of recent meta-analyses. Ther. Adv. Psychopharmacol. 2018, 8, 303–318.
- Leucht, S.; Cipriani, A.; Spineli, L.; Marvidis, D.; Örey, D.; Richter, F.; Samara, M.; Barbui, C.; Engel, R.R.; Geddes, J.R.; et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: A multiple-treatments meta-analysis. Lancet 2013, 382, 951–962.
- Elkis, H. Treatment-resistant schizophrenia. Psychiatr. Clin. N. Am. 2007, 30, 511–533.
- Kane, J.; Honigfeld, G.; Singer, J.; Meltzer, H. Clozapine for the treatment-resistant schizophrenic. Arch. Gen. Psychiatry 1988, 45, 789–796.
- Rosenheck, R.; Cramer, J.; Xu, W.; Thomas, J.; Henderson, W.; Frisman, L.; Fye, C.; Charney, D. A Comparison of clozapine and haloperidol in hospitalized patients with Refractory Schizophrenia. N. Engl. J. Med. 1997, 337, 809–815.
- Siskind, D.; Siskind, V.; Kisely, S. Clozapine Response Rates among People with Treatment-Resistant Schizophrenia: Data from a Systematic Review and Meta-Analysis. Can. J. Psychiatry 2017, 62, 772–777.
- Blackman, G.M.; Lisshammar, J.E.M.; Zafar, R.M.; Pollak, T.A.; Pritchard, M.M.; Cullen, A.E.; Rogers, J.M.B.; Carter, B.; Griffiths, K.M.; Nour, M.B.B.; et al. Clozapine Response in Schizophrenia and Hematological Changes. J. Clin. Psychopharmacol. 2020, 41, 19–24.
- Stilo, S.A.; Murray, R.M. Non-Genetic Factors in Schizophrenia. Curr. Psychiatry Rep. 2019, 21, 100.
- Trubetskoy, V.; Pardiñas, A.F.; Qi, T.; Panagiotaropoulou, G.; Awasthi, S.; Bigdeli, T.B.; Bryois, J.; Chen, C.-Y.; Dennison, C.A.; Hall, L.S.; et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 2022, 604, 502–508.
- Kushima, I.; Nakatochi, M.; Aleksic, B.; Okada, T.; Kimura, H.; Kato, H.; Morikawa, M.; Inada, T.; Ishizuka, K.; Torii, Y.; et al. Cross-Disorder Analysis of Genic and Regulatory Copy Number Variations in Bipolar Disorder, Schizophrenia, and Autism Spectrum Disorder. Biol. Psychiatry 2022, 92, 362–374.
- Kushima, I.; Aleksic, B.; Nakatochi, M.; Shimamura, T.; Okada, T.; Uno, Y.; Morikawa, M.; Ishizuka, K.; Shiino, T.; Kimura, H.; et al. Comparative Analyses of Copy-Number Variation in Autism Spectrum Disorder and Schizophrenia Reveal Etiological Overlap and Biological Insights. Cell Rep. 2018, 24, 2838–2856.
- Kushima, I.; Aleksic, B.; Nakatochi, M.; Shimamura, T.; Shiino, T.; Yoshimi, A.; Kimura, H.; Takasaki, Y.; Wang, C.; Xing, J.; et al. High-resolution copy number variation analysis of schizophrenia in Japan. Mol. Psychiatry 2016, 22, 430–440.
- Sarowar, T.; Grabrucker, A.M. Rho GTPases in the Amygdala—A Switch for Fears? Cells 2020, 9, 1972.
- Duman, J.G.; Blanco, F.A.; Cronkite, C.A.; Ru, Q.; Erikson, K.C.; Mulherkar, S.; Bin Saifullah, A.; Firozi, K.; Tolias, K.F. Rac-maninoff and Rho-vel: The symphony of Rho-GTPase signaling at excitatory synapses. Small GTPases 2021, 13, 14–47.
- Huang, G.-H.; Sun, Z.-L.; Li, H.-J.; Feng, D.-F. Rho GTPase-activating proteins: Regulators of Rho GTPase activity in neuronal development and CNS diseases. Mol. Cell. Neurosci. 2017, 80, 18–31.
- Ramos-Miguel, A.; Barr, A.M.; Honer, W.G. Spines, synapses, and schizophrenia. Biol. Psychiatry 2015, 78, 741–743.
- Mould, A.W.; Al-Juffali, N.; von Delft, A.; Brennan, P.E.; Tunbridge, E.M. Kalirin as a Novel Treatment Target for Cognitive Dysfunction in Schizophrenia. CNS Drugs 2021, 36, 1–16.
- Hanifa, M.; Singh, M.; Randhawa, P.K.; Jaggi, A.S.; Bali, A. A focus on Rho/ROCK signaling pathway: An emerging therapeutic target in depression. Eur. J. Pharmacol. 2023, 946, 175648.
- Van Bokhoven, H. Genetic and epigenetic networks in intellectual disabilities. Annu. Rev. Genet. 2011, 45, 81–104.
- Liaci, C.; Camera, M.; Caslini, G.; Rando, S.; Contino, S.; Romano, V.; Merlo, G.R. Neuronal Cytoskeleton in Intellectual Disability: From Systems Biology and Modeling to Therapeutic Opportunities. Int. J. Mol. Sci. 2021, 22, 6167.
- Guo, D.; Yang, X.; Shi, L. Rho GTPase Regulators and Effectors in Autism Spectrum Disorders: Animal Models and Insights for Therapeutics. Cells 2020, 9, 835.
- Mosaddeghzadeh, N.; Ahmadian, M.R. The RHO Family GTPases: Mechanisms of Regulation and Signaling. Cells 2021, 10, 1831.
- Symons, M.; Settleman, J. Rho family GTPases: More than simple switches. Trends Cell Biol. 2000, 10, 415–419.
- Cherfils, J.; Zeghouf, M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013, 93, 269–309.
- Jaiswal, M.; Dvorsky, R.; Ahmadian, M.R. Deciphering the molecular and functional basis of Dbl family proteins: A novel systematic approach toward classification of selective activation of the Rho family proteins. J. Biol. Chem. 2013, 288, 4486–4500.
- Kreider-Letterman, G.; Carr, N.M.; Garcia-Mata, R. Fixing the GAP: The role of RhoGAPs in cancer. Eur. J. Cell Biol. 2022, 101, 151209.
- Bustelo, X.R.; Sauzeau, V.; Berenjeno, I.M. GTP-binding proteins of the Rho/Rac family: Regulation, effectors and functions in vivo. BioEssays 2007, 29, 356–370.
- Amin, E.; Dubey, B.N.; Zhang, S.-C.; Gremer, L.; Dvorsky, R.; Moll, J.M.; Taha, M.S.; Nagel-Steger, L.; Piekorz, R.P.; Somlyo, A.V.; et al. Rho-kinase: Regulation, (dys)function, and inhibition. Biol. Chem. 2013, 394, 1399–1410.
- Amano, M.; Kanazawa, Y.; Kozawa, K.; Kaibuchi, K. Identification of the Kinase-Substrate Recognition Interface between MYPT1 and Rho-Kinase. Biomolecules 2022, 12, 159.
- Grassie, M.E.; Moffat, L.D.; Walsh, M.P.; MacDonald, J.A. The myosin phosphatase targeting protein (MYPT) family: A regulated mechanism for achieving substrate specificity of the catalytic subunit of protein phosphatase type 1delta. Arch. Biochem. Biophys. 2011, 510, 147–159.
- Seccia, T.M.; Rigato, M.; Ravarotto, V.; Calò, L.A. ROCK (RhoA/Rho Kinase) in Cardiovascular–Renal Pathophysiology: A Review of New Advancements. J. Clin. Med. 2020, 9, 1328.
- Luo, L. RHO GTPASES in neuronal morphogenesis. Nat. Rev. Neurosci. 2000, 1, 173–180.
- Civiero, L.; Greggio, E. PAKs in the brain: Function and dysfunction. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2018, 1864, 444–453.
- Dickson, B.J. Rho GTPases in growth cone guidance. Curr. Opin. Neurobiol. 2001, 11, 103–110.
- Burridge, K.; Guilluy, C. Focal adhesions, stress fibers and mechanical tension. Exp. Cell Res. 2015, 343, 14–20.
- Benarroch, E.E. Rho GTPases: Role in dendrite and axonal growth, mental retardation, and axonal regeneration. Neurology 2007, 68, 1315–1318.
- Newey, S.E.; Velamoor, V.; Govek, E.-E.; Van Aelst, L. Rho GTPases, dendritic structure, and mental retardation. J. Neurobiol. 2005, 64, 58–74.
- Sekiguchi, M.; Sobue, A.; Kushima, I.; Wang, C.; Arioka, Y.; Kato, H.; Kodama, A.; Kubo, H.; Ito, N.; Sawahata, M.; et al. ARHGAP10, which encodes Rho GTPase-activating protein 10, is a novel gene for schizophrenia risk. Transl. Psychiatry 2020, 10, 247.
- Guo, W.; Cai, Y.; Zhang, H.; Yang, Y.; Yang, G.; Wang, X.; Zhao, J.; Lin, J.; Zhu, J.; Li, W.; et al. Association of ARHGAP18 polymorphisms with schizophrenia in the Chinese-Han population. PLoS ONE 2017, 12, e0175209.
- Potkin, S.G.; Turner, J.A.; Fallon, J.A.; Lakatos, A.; Keator, D.B.; Guffanti, G.; Macciardi, F. Gene discovery through imaging genetics: Identification of two novel genes associated with schizophrenia. Mol. Psychiatry 2008, 14, 416–428.
- Potkin, S.G.; Macciardi, F.; Guffanti, G.; Fallon, J.H.; Wang, Q.; Turner, J.A.; Lakatos, A.; Miles, M.F.; Lander, A.; Vawter, M.P.; et al. Identifying gene regulatory networks in schizophrenia. NeuroImage 2010, 53, 839–847.
- Ohi, K.; Hashimoto, R.; Nakazawa, T.; Okada, T.; Yasuda, Y.; Yamamori, H.; Fukumoto, M.; Umeda-Yano, S.; Iwase, M.; Kazui, H.; et al. The p250GAP gene is associated with risk for schizophrenia and schizotypal personality traits. PLoS ONE 2012, 7, e35696.
- Gandal, M.J.; Zhang, P.; Hadjimichael, E.; Walker, R.L.; Chen, C.; Liu, S.; Won, H.; Van Bakel, H.; Varghese, M.; Wang, Y.; et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 2018, 362, eaat8127.
- Kushima, I.; Nakamura, Y.; Aleksic, B.; Ikeda, M.; Ito, Y.; Shiino, T.; Okochi, T.; Fukuo, Y.; Ujike, H.; Suzuki, M.; et al. Resequencing and association analysis of the KALRN and EPHB1 genes and their contribution to schizophrenia susceptibility. Schizophr. Bull. 2010, 38, 552–560.
- Russell, T.A.; Grubisha, M.J.; Remmers, C.L.; Kang, S.K.; Forrest, M.P.; Smith, K.R.; Kopeikina, K.J.; Gao, R.; Sweet, R.A.; Penzes, P. A Schizophrenia-Linked KALRN Coding Variant Alters Neuron Morphology, Protein Function, and Transcript Stability. Biol. Psychiatry 2018, 83, 499–508.
- Gulsuner, S.; Stein, D.J.; Susser, E.S.; Sibeko, G.; Pretorius, A.; Walsh, T.; Majara, L.; Mndini, M.M.; Mqulwana, S.G.; Ntola, O.A.; et al. Genetics of schizophrenia in the South African Xhosa. Science 2020, 367, 569–573.
- Mizuki, Y.; Takaki, M.; Okahisa, Y.; Sakamoto, S.; Kodama, M.; Ujike, H.; Uchitomi, Y. Human Rho guanine nucleotide exchange factor 11 gene is associated with schizophrenia in a Japanese population. Hum. Psychopharmacol. Clin. Exp. 2014, 29, 552–558.
- Kimura, H.; Fujita, Y.; Kawabata, T.; Ishizuka, K.; Wang, C.; Iwayama, Y.; Okahisa, Y.; Kushima, I.; Morikawa, M.; Uno, Y.; et al. A novel rare variant R292H in RTN4R affects growth cone formation and possibly contributes to schizophrenia susceptibility. Transl. Psychiatry 2017, 7, e1214.
- Hsu, R.; Woodroffe, A.; Lai, W.-S.; Cook, M.N.; Mukai, J.; Dunning, J.P.; Swanson, D.J.; Roos, J.L.; Abecasis, G.R.; Karayiorgou, M.; et al. Nogo Receptor 1 (RTN4R) as a candidate gene for schizophrenia: Analysis using human and mouse genetic approaches. PLoS ONE 2007, 2, e1234.
- Sinibaldi, L.; De Luca, A.; Bellacchio, E.; Conti, E.; Pasini, A.; Paloscia, C.; Spalletta, G.; Caltagirone, C.; Pizzuti, A.; Dallapiccola, B. Mutations of the Nogo-66 receptor (RTN4R) gene in schizophrenia. Hum. Mutat. 2004, 24, 534–535.
- McCarthy, S.E.; Makarov, V.; Addington, A.M.; McClellan, J.; Yoon, S.; Perkins, D.O.; Dickel, D.E.; Kusenda, M.; Krastoshevsky, O.; Krause, V.; et al. Microduplications of 16p11.2 are associated with schizophrenia. Nat. Genet. 2009, 41, 1223–1227.
- Golzio, C.; Willer, J.; Talkowski, M.E.; Oh, E.C.; Taniguchi, Y.; Jacquemont, S.; Reymond, A.; Sun, M.; Sawa, A.; Gusella, J.F.; et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature 2012, 485, 363–367.
- Shibata, H.; Oishi, K.; Yamagiwa, A.; Matsumoto, M.; Mukai, H.; Ono, Y. PKNbeta interacts with the SH3 domains of Graf and a novel Graf related protein, Graf2, which are GTPase activating proteins for Rho family. J. Biochem. 2001, 130, 23–31.
- Hada, K.; Wulaer, B.; Nagai, T.; Itoh, N.; Sawahata, M.; Sobue, A.; Mizoguchi, H.; Mori, D.; Kushima, I.; Nabeshima, T.; et al. Mice carrying a schizophrenia-associated mutation of the Arhgap10 gene are vulnerable to the effects of methamphetamine treatment on cognitive function: Association with morphological abnormalities in striatal neurons. Mol. Brain 2021, 14, 21.
- Maeda, M.; Hasegawa, H.; Hyodo, T.; Ito, S.; Asano, E.; Yuang, H.; Funasaka, K.; Shimokata, K.; Hasegawa, Y.; Hamaguchi, M.; et al. ARHGAP18, a GTPase-activating protein for RhoA, controls cell shape, spreading, and motility. Mol. Biol. Cell 2011, 22, 3840–3852.
- Thompson, W.R.; Yen, S.S.; Uzer, G.; Xie, Z.; Sen, B.; Styner, M.; Burridge, K.; Rubin, J. LARG GEF and ARHGAP18 orchestrate RhoA activity to control mesenchymal stem cell lineage. Bone 2017, 107, 172–180.
- Humphries, B.; Wang, Z.; Li, Y.; Jhan, J.-R.; Jiang, Y.; Yang, C. ARHGAP18 Downregulation by miR-200b Suppresses Metastasis of Triple-Negative Breast Cancer by Enhancing Activation of RhoA. Cancer Res. 2017, 77, 4051–4064.
- Nakazawa, T.; Watabe, A.M.; Tezuka, T.; Yoshida, Y.; Yokoyama, K.; Umemori, H.; Inoue, A.; Okabe, S.; Manabe, T.; Yamamoto, T. p250GAP, a novel brain-enriched GTPase-activating protein for Rho family GTPases, is involved in the N-Methyl-d-aspartate receptor signaling. Mol. Biol. Cell 2003, 14, 2921–2934.
- Nakazawa, T.; Kuriu, T.; Tezuka, T.; Umemori, H.; Okabe, S.; Yamamoto, T. Regulation of dendritic spine morphology by an NMDA receptor-associated Rho GTPase-activating protein, p250GAP. J. Neurochem. 2008, 105, 1384–1393.
- Kannan, M.; Lee, S.-J.; Schwedhelm-Domeyer, N.; Nakazawa, T.; Stegmüller, J. p250GAP is a novel player in the Cdh1-APC/Smurf1 pathway of axon growth regulation. PLoS ONE 2012, 7, e50735.
- Paskus, J.D.; Herring, B.E.; Roche, K.W. Kalirin and Trio: RhoGEFs in Synaptic Transmission, Plasticity, and Complex Brain Disorders. Trends Neurosci. 2020, 43, 505–518.
- Penzes, P.; Johnson, R.C.; Kambampati, V.; Mains, R.E.; Eipper, B.A. Distinct roles for the two Rho GDP/GTP exchange factor domains of kalirin in regulation of neurite growth and neuronal morphology. J. Neurosci. 2001, 21, 8426–8434.
- Yan, Y.; Eipper, B.A.; Mains, R.E. Kalirin-9 and Kalirin-12 Play Essential Roles in Dendritic Outgrowth and Branching. Cereb. Cortex 2014, 25, 3487–3501.
- Deo, A.J.; Cahill, M.E.; Li, S.; Goldszer, I.; Henteleff, R.; VanLeeuwen, J.-E.; Rafalovich, I.; Gao, R.; Stachowski, E.K.; Sampson, A.R.; et al. Increased expression of Kalirin-9 in the auditory cortex of schizophrenia subjects: Its role in dendritic pathology. Neurobiol. Dis. 2012, 45, 796–803.
- Grubisha, M.J.; Sun, T.; Eisenman, L.; Erickson, S.L.; Chou, S.-Y.; Helmer, C.D.; Trudgen, M.T.; Ding, Y.; Homanics, G.E.; Penzes, P.; et al. A Kalirin missense mutation enhances dendritic RhoA signaling and leads to regression of cortical dendritic arbors across development. Proc. Natl. Acad. Sci. USA 2021, 118, e2022546118.
- Harrington, A.W.; Li, Q.M.; Tep, C.; Park, J.B.; He, Z.; Yoon, S.O. The role of Kalirin9 in p75/nogo receptor-mediated RhoA activation in cerebellar granule neurons. J. Biol. Chem. 2008, 283, 24690–24697.
- Yamashita, T.; Tucker, K.L.; Barde, Y.-A. Neurotrophin binding to the p75 receptor modulates Rho activity and axonal outgrowth. Neuron 1999, 24, 585–593.
- Boghdadi, A.G.; Teo, L.; Bourne, J.A. The Involvement of the Myelin-Associated Inhibitors and Their Receptors in CNS Plasticity and Injury. Mol. Neurobiol. 2017, 55, 1831–1846.
- Rümenapp, U.; Blomquist, A.; Schwörer, G.; Schablowski, H.; Psoma, A.; Jakobs, K.H. Rho-specific binding and guanine nucleotide exchange catalysis by KIAA0380, a dbl family member. FEBS Lett. 1999, 459, 313–318.
- Fukuhara, S.; Murga, C.; Zohar, M.; Igishi, T.; Gutkind, J.S. A Novel PDZ domain containing guanine nucleotide exchange factor links heterotrimeric G proteins to Rho. J. Biol. Chem. 1999, 274, 5868–5879.
- Jackson, M.; Song, W.; Liu, M.-Y.; Jin, L.; Dykes-Hoberg, M.; Lin, C.-L.G.; Bowers, W.J.; Federoff, H.J.; Sternweis, P.C.; Rothstein, J.D. Modulation of the neuronal glutamate transporter EAAT4 by two interacting proteins. Nature 2001, 410, 89–93.
- Mizuki, Y.; Takaki, M.; Sakamoto, S.; Okamoto, S.; Kishimoto, M.; Okahisa, Y.; Itoh, M.; Yamada, N. Human Rho Guanine Nucleotide Exchange Factor 11 (ARHGEF11) Regulates Dendritic Morphogenesis. Int. J. Mol. Sci. 2016, 18, 67.
- Mizuki, Y.; Sakamoto, S.; Okahisa, Y.; Yada, Y.; Hashimoto, N.; Takaki, M.; Yamada, N. Mechanisms Underlying the Comorbidity of Schizophrenia and Type 2 Diabetes Mellitus. Int. J. Neuropsychopharmacol. 2020, 24, 367–382.
- Davidkova, G.; Mccullumsmith, R.E.; Meador-Woodruff, J.H. Expression of ARHGEF11 mRNA in schizophrenic thalamus. Ann. N. Y. Acad. Sci. 2003, 1003, 375–377.
- Karayiorgou, M.; Simon, T.J.; Gogos, J.A. 22q11.2 microdeletions: Linking DNA structural variation to brain dysfunction and schizophrenia. Nat. Rev. Neurosci. 2010, 11, 402–416.
- Schwab, M.E. Functions of Nogo proteins and their receptors in the nervous system. Nat. Rev. Neurosci. 2010, 11, 799–811.
- Perlstein, M.D.; Chohan, M.R.; Coman, I.L.; Antshel, K.M.; Fremont, W.P.; Gnirke, M.H.; Kikinis, Z.; Middleton, F.A.; Radoeva, P.D.; Shenton, M.E.; et al. White matter abnormalities in 22q11.2 deletion syndrome: Preliminary associations with the Nogo-66 receptor gene and symptoms of psychosis. Schizophr. Res. 2014, 152, 117–123.
- Fernandez-Enright, F.; Andrews, J.L.; Newell, K.A.; Pantelis, C.; Huang, X.F. Novel implications of Lingo-1 and its signaling partners in schizophrenia. Transl. Psychiatry 2014, 4, e348.
- Jitoku, D.; Hattori, E.; Iwayama, Y.; Yamada, K.; Toyota, T.; Kikuchi, M.; Maekawa, M.; Nishikawa, T.; Yoshikawa, T. Association study of Nogo-related genes with schizophrenia in a Japanese case-control sample. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2011, 156, 581–592.
- Lin, G.N.; Corominas, R.; Lemmens, I.; Yang, X.; Tavernier, J.; Hill, D.E.; Vidal, M.; Sebat, J.; Iakoucheva, L.M. Spatiotemporal 16p11.2 protein network implicates cortical late mid-fetal brain development and KCTD13-Cul3-RhoA pathway in psychiatric diseases. Neuron 2015, 85, 742–754.
- Chen, Y.; Yang, Z.; Meng, M.; Zhao, Y.; Dong, N.; Yan, H.; Liu, L.; Ding, M.; Peng, H.B.; Shao, F. Cullin mediates degradation of RhoA through evolutionarily conserved BTB adaptors to control actin cytoskeleton structure and cell movement. Mol. Cell 2009, 35, 841–855.
- Genschik, P.; Sumara, I.; Lechner, E. The emerging family of CULLIN3-RING ubiquitin ligases (CRL3s): Cellular functions and disease implications. EMBO J. 2013, 32, 2307–2320.
- Urresti, J.; Zhang, P.; Moran-Losada, P.; Yu, N.-K.; Negraes, P.D.; Trujillo, C.A.; Antaki, D.; Amar, M.; Chau, K.; Pramod, A.B.; et al. Cortical organoids model early brain development disrupted by 16p11.2 copy number variants in autism. Mol. Psychiatry 2021, 26, 7560–7580.