During tissue turnover, the telomeres of cells undergoing differentiation can be damaged as a consequence of defective DNA repair caused by endogenous or exogenous agents. This may result in the emergence of new mechanism of telomere maintenance which is the final outcome of DNA damage and the initial signal that triggers malignant transformation. Instead, transformation of stem cells is directly induced by primary derangement of telomere maintenance mechanisms. The newly modified telomere complex may promote survival of cancer stem cells, independently of telomere maintenance. An inherent resistance of stem cells to transformation may be linked to specific, robust mechanisms that help maintain telomere integrity.
- DNA damage response (DDR)
- DNA damage
1.
Introduction
stem cells are endowed with a specific mechanism of telomere maintenance which is lost as son or shortly after they leave the stem cell compartment. It is is essential for their indefinite capacity of regeneration, an ability also shared by cancer cells. However, the immunophenotype of a vast majority of cancers display markers of differentiation indicative of an origin at different stages of development posterior to the stem cell stage. This suggests that carconogenesis implies the gain of a stem-like mechanism of telomere maintenance by committed cells.
Most, if not all, common adult cancers exhibit markers of differentiation corresponding to distinct differentiation stages. (1,2)Concurrently, their telomeres are stablized but are reduced in lenght compared to normal counterparts, again suggesting that common adult cancers arise from cells that, even if they are not terminally differentiated, are in the process of differentiation and concomitant telomere erosion. Furthermore, the carcinoma in situ stage, a typical feature of common adult cancers, seems to be reached at a critical threshold of telomere erosion
On the contrary, tumors exhibiting an undifferentiated/immature phenotype and sudden onset that apparently precludes sustained gradual telomere erosion, such as early childhood cancers, acute leukemia and some sarcomas, do not appear to develop through a carcinoma in situ stage, but even in these tumors a substantial degree of telomere erosion is usually observed [3,4], suggesting a former process of telomere length reduction before malignant transformation stabilizes telomere length. The lack of an in situ stage could be attributed to failure of detection due to rapid growth of these tumor but could, as well, be an inherent property of these tumors which could not require a periodo of telomere erosion and would be endowed with indefinte self-renewal from their inception.
Interference with the telomere-maintenance-specific mechanism/s of stem cells may be a feature of these tumors. For instance, it has been shown that c-Myc overexpression within the stem cell compartment results in cellular exit from the stem cell compartment and increased differentiation at the expense of self-renewal [5]. At the same time, progenitors that leave the stem cell compartment as a result of c-myc overexpression become vulnerable to carcinogenesis. This may be associated with the impact of c-Myc on exit from the stem cell pool and probably interference with this telomere maintenance mechanism/s specific to stem cells due to the ability of c-Myc to activate hTERT expression [6]. C-Myc has been shown to induce different secondary oncogenic targets depending on the stage of differentiation of the responding cell. For instance, Eµ-c-Myc transgenic mice develop B-cell lymphomas of both mature and immature origin. Nepal et al. (7] showed that 90% of mature B-cell lymphomas (B220+ sIg+ ) overexpressed Mdm2, Arf, or p53 either alone or in combination, whereas none of the evaluated immature lymphomas in Eµ-c-Myc mice overexpressed Arf or p53 and only 27% overexpressed Mdm2. Western blot analysis of the c-myc-induced apoptotic pathway in the immature B-cell lymphomas revealed that none of the six evaluated tumors analyzed showed accumulation of Arf, Mdm2, or p53. These data indicate that c-myc induces the Arf-p53 pathway in mature B cells but not in pro- and pre-B cells [7]. Thus, although p53 activation is functional throughout B-cell development, its inactivation seems crucial only for mature, but not immature, tumors [7]. There is a parallelism with the concept of two different functions of p53 at early and late phases of hematopoietic development as it has been reported that in the hematopoietic stem cell compartment, p53 regulates not apoptosis, but stem cell quiescence and exit from the stem cell pool through its targets Necdin and Gf-1 and independently of p21(8,9) The same concept is supported by specific knock-in p53 mutations that display different protective effects in mature and immature lymphomas [10,11)
Information gathered on therapy using human-induced pluripotent stem cells (hiPSCs) revealed that tumorigenicity caused by reprogramming factors Oct3/4, Sox2, Klf4, and cMyc should be mainly attributed to cMyc reactivation. In Yamanaka`s words, “chimeric mice made with iPSCs created by the induction of retrovirus-mediated transfection of the four reprogramming factors often developed tumors”. We detected reactivation of c-Myc retrovirus in these tumors. Chimeric mice with iPSCs that had not been induced with the c-Myc retrovirus did not show such tumors. Another subset of tumors and teratomas formed by transplanted iPSCs may be related to faulty differentiation–expansion of progenitors in the absence of a physiological microenvironment, as shown by the fact that efficient methods of in vitro-directed differentiation reduce the risk of teratoma and tumor formation [12].
Previous surveys have highlighted a difference in cancer pathways between tumors of putative stem cell origin and more differentiated/mature tumors: a wide spectrum of diverse cancer-driving mutations that enhance proliferation/decrease apoptosis is usually identified in common adult/mature tumors, whereas pathways that can directly affect telomere maintenance drove transformation of true stem cell/early progenitor tumors. One instance taken from the human clinic in support of this contention is the Beckwith–Wiedemann syndrome, a genetic disease of the telomerase reverse transcriptase (TERT)-TGFβ-Smad pathway which is associated with an 800-fold risk of developing childhood cancer in many different organs [13]. Another is represented by the tumor model reported by Passegué et al. and Santaguida et al., in which JunB inactivation specifically expanded the number of long-term hematopoietic stem cells (LT-HDCs) leading to myeloproliferative disease [14,15]. Since the growth-promoting effects of telomerase have been linked to TGFβ inhibition and the expression of the TGFβ effector gene JunB is decreased in mice that overexpress mTert (16) it is apparent that the same, or closely related, pathway as that of Beckwith–Wiedemann syndrome may be involved in this tumor models. One way to unify the cancer pathways of mature and stem/early progenitors is to hypothesize that the common outcomes of the independent mutations that are prevalent in common adult cancers may converge in a final step of telomere attrition/dysfunction, which, similar to the crisis observed in in vitro culture, might trigger reactivation of telomerase or ALT which is indispensable for the indefinite survival of cancer cells. Instead, tumors of stem cell origin appear to be driven by pathways that directly impact the telomere maintenance mechanism/s.
A prediction of this hypothesis is that most if not all oncogenic agents should be able to alter the telomere complex
- Review and Hypothesis
The DNA Damage Response. Telomeres and cancer
1.
We hypothesize that most carcinogenic pathways provoke or are associated with an altered DNA damage response which, through disruption of checkpoint control or other mechanisms that are not completely understood, disturb telomere homeostasis and thereby induce the emergence of new anomalous mechanisms of telomere maintenance
A perusal of the literature shows that deregulation of most DNA damage response components is concurrently associated with both telomere dysfunction/erosion and cancer predisposition (Tables 1 and 2).
Table 1. Components of the DNA damage response responsible for telomere dysfunction and cancer.
|
|
Telomere Dysfunction |
Cancer Predisposition |
|
* P53 |
Yes |
Yes |
|
Rad 9 |
Yes |
Yes |
|
Rad 51 |
Yes |
Yes |
|
Rad 52 |
Yes |
Yes |
|
Rad 54 |
Yes |
Yes |
|
c-Abl |
Yes |
Yes |
|
BRCA1-BRCA2 |
Yes |
Yes |
|
** PARP-1 |
Yes |
Yes |
|
*** DNA-PKcs |
Yes |
Yes |
|
Artemis |
Yes |
Yes |
|
Cerunnos |
Yes |
Yes |
|
Ku70-Ku80 |
Yes |
Yes |
|
Ligase IV |
Yes |
Yes |
|
Fanconi Anemia |
Yes |
Yes |
|
^ CtIP |
Yes |
Yes |
|
Rad17 |
Yes |
Yes |
|
9-1-1 complex |
Yes |
Yes |
|
14-3-3 complex |
Yes |
Yes |
|
CTC complex |
Yes |
Yes |
Table 2. Components of the DDR which are responsible for DNA repair syndromes that manifest early in life as clinical entities and are concurrently associated with cancer predisposition and telomere homeostasis.
|
|
Telomere Dysfunction |
Cancer Predisposition |
|
(See comment) ERCC6like2 (ERCC6L2) |
Yes |
Yes |
|
CMMRD-Xeroderma pigmentosum |
Yes |
Yes |
|
Bloom syndrome |
Yes |
Yes |
|
Werner syndrome |
Yes |
Yes |
|
Rothmund–Thomson syndrome |
Yes [ |
Yes |
See these tables with notes and references in (17)
- False Exceptions to the Correlation between Cancer–Telomere Dysfunction: Seckel Syndrome, Cockayne Syndrome, Trichothiodystrophy
In humans, Seckel syndrome type 1 is caused by an autosomal recessive mutation in ATR. Patients with this condition exhibit developmental defects but no cancer predisposition. Nevertheless, an autosomal dominant germline ATR missense mutation associated with oropharyngeal cancer was found in 24 individuals from a five-generation pedigree. Loss of heterozygosity was observed in tumor tissue( 18). There are observations suggesting that DNA repair in Seckel syndrome is aborted in its initial stages, which may conceivably lead to arrested proliferation or be incompatible with cell life owing to ATR loss of function [19]. Dramatic proliferation failure of ATR-deficient cells is observed in vivo as well as in in vitro cell culture, apparently due to the lack of stabilization of replication forks in the absence or deficiency of ATR. The rapid exhaustion of replication ability in ATR-deficient cells may explain the rarity of tumors either in ATR null mice or in human Seckel syndrome [20] or even the small occurrence of tumors in heterozygous mice suggesting that unrepaired DNA is incompatible with cell lif.
Cockayne syndrome (CS) is caused by a specific failure in the mechanism of transcription-coupled repair due to defects in either of the two genes ERCC6 (CSB) or ERCC8 (CSA). Thse defects lead to inhibition of RNA polI in numerous genes (either active or inactive gnes) Functionally, the end result appears to be analogous to that of Seckel syndrome in that interference with RNA transcription can make the cell unable to proliferate and execute DNA repair (21,22) As in the case of Seckel syndrome, this can be incompatible with cancer transformation.
A similar mechanism could explain the lack of cancer predisposition in trichothiodystrophy (TTD). TTD is caused by TTDN1, a gene of unknown function or by TTDA which encodes a small subunit of TFIIH. THIIH has a vital function in the initiation of RNA transcription [21,22)].
- Mutation of the Mre11 Component of the MRN Complex Does Not Result in Telomere Erosion nor Cancer Predisposition
Germline alterations of MRN components give rise to hereditary cancer predisposition syndromes ataxia-telangiectasia like disease (A-TLD) and Nijmegen breakage syndrome (NBS), caused by hippomorphic mutation in the gene encoding Mre11 nuclease and NBS1 (encoding the protein NBN also known as nibrin) (null mutation of Nbs1 causes embryonic lethality). These syndromes are phenotypically similar to ataxia-telangiectasia, (AT), a hereditary cancer predisposition syndrome caused by ATM deficiency which presents with cerebellar degeneration, immunodeficiency, reduced fertility, radiosensitivity, and cancer predisposition. Similar phenotypic features are shared by Nijmegen breakage syndrome (NBS) except that it presents with microcephaly in place of cerebellar degeneration and A-TLD, although the latter may have a milder clinical expression. Cancer predisposition of AT and NBS is conspicuous and manifests mainly in lymphomas related to chromosomal aberrations at the sites of T-cell receptor or immunoglobulin chain rearrangement where V(D)J recombination spontaneously induces DSBs. However, other cancer types may occur. (23,24). A feature displayed by the Mre11ATLD/ATLD mutation might explain the different tumor-promoting activities of MRN component Mre11 versus Nbs1 and ATM. Shortened telomeres associate with defective ATM or Nbs1 function [25], but telomere shortening is absent in cells homozygous for Mre11ATLD/ATLD. Attwool et al. reported that no telomere attrition was seen in immortalized Mre11ATLD/ATLD MEFs over the course of over 300 population doublings (26)
Spehalski et al (27) introduced the Mre11H129N mutation in B lymphocytes through CD19-Cre expresión. This,did not affect the survival of p53 null mice and thymic lymphomas typical of p53 deficiency developed with the same frequency regardless of Mre11 deficiency.
Next, these authors examined the impact of Mre11 deficiency on the Artemis mouse model strongly predisposed to B lineage lymphomas. Artemis is required for processing the DNA ends generated during V(D)J recombination. Mutations in Artemis cause immunodeficiencies and tumors. When combined with p53 deficiency, aggressive pro-B lymphomas harboring chromosomal translocations involving IgH and c-Myc or N-Myc loci develop. The majority of Artemis/p53 double nulls succumbed to progenitor B lymphoma and a smaller subset of thymic lymphoma of progenitor T-cell origin. Remarkably, no pro-B-cell lymphoma arose in Artemis/p53 double null mice with either Mre11−/− (n = 11) or Mre11−/H129N. The majority of lymphomas that developed in these mice were thymic lymphomas. These results indicate that the Mre11 mutation suppresses pro-B-cell lymphomas in a context where pro-B-cell lymphomas normally arise in the majority of mice by 8–10 weeks of age.
In this context it may be pertinente to take into consideration a former report which showed that development of thymic lymphoma in p53-deficient mice does not require V(D)J recombination (28). This may suggest that Artemis mutation that impairs V(D)J recombination is rescued by the telomere preservation associated to Mre11 mutation whereas Mre11 mutation could not achieve the same telomere effect when brought about by p53
Table 3. Common and differential features of ataxia-telangiectasia mutated, ataxia-telangiectasis-like disease, and Nijmegen breakage syndrome.
|
ATM-Mutated |
Mer11 (AT-LD) |
NBN (Nijmegen Breakage Syndrome) (NBS) |
|
Deficient initial DNA repair. |
Deficient initial DNA repair. |
Deficient initial DNA repair. |
|
Hypersensitivity to IR-induced chromosome breakage, increased translocations (chromosomal instability). |
Hypersensitivity to IR-induced chromosome breakage, increased translocations (chromosomal instability). |
Hypersensitivity to IR-induced chromosome breakage, increased translocations (chromosomal instability). |
|
IR-induced thymocyte apoptosis relative to wild-type: 58% versus 90%. Severe intra-S and G2/M checkpoint defects. Telomere shortening. Cancer predisposition. |
IR-induced thymocyte apoptosis relative to wild-type: Sightly reduced (81% versus 90%. Intra-S and G2/M checkpoints less severe than in ATM deficiency and comparable to Nsb1 null. No telomere shortening. No cancer predisposition (or mild). |
IR-induced thymocyte apoptosis: Extreme variations in apoptotic capacity (in neural tissue similar to wild-type). Intra-S and G2/M checkpoints less severe than in ATM deficiency and comparable to Mre11 hypomorphic. Telomere shortening. Cancer predisposition. |
- Replication Stress is associated with Cancer
Transformation
Replication stress (RS) refers to disturbances in DNA dynamics that are reflected in the slowing and asymmetry of replication fork progression. It also affects deregulation of replication origins. There are multiple causes of replication stress, including oncogenes, shortage of nucleotides and replication factors, UV or ionizing radiation, and reactive oxygen species, as well as difficulties arising in the process of DNA replication due to special features of particular DNA regions, such as repetitive elements, late replication or anomalous chromatin configuration. Some of these causes have been shown to be interrelated, such as oncogenes and exhaustion of nucleotide pools. Nucleotide deficiency has been shown to promote genomic instability in early stages of tumor development (29,30) More recently, Tummala et al (31) have reported individuals with clinical features of the cancer-predisposition syndrome dyskeratosis congénita (DC) that harboured los of function mutation of TYMS gene(thymidylate synthase) combined with an epistatic effect from its antisensense regulator. This represents a new molecular pathway for DC. TYMS-ENOSF1 defects lead to decreased deoxyribonucleotide pools, ribonucleotide reductase inhibition and a DNA damage response that affects expresión of proteins involved in telomere maintenance.
Tolerance to low levels of replication stress or defects in checkpoint and mitotic control resulting from deficient resolution of chromatid entanglement can easily lead to the loss of DNA sequences. In common fragile sites (CFSs), this may underlie the frequent inactivation of tumor suppressors in these regions (30) while in telomeres it will likely be reflected by changes in length and/or telomere dysfunction, whereas loss of centromeric or ribosomal DNA will most likely lead to impaired cell function or even cell death. We hypothesize that loss of tumor suppressors or mutations in genomic DNA can enhance cell proliferation, which cannot result in transformation unless a new telomere maintenance mechanism arises. Telomere dysfunction can also result in cell arrest or cell death, but in some cases may lead to escape of cell death through different mechanisms that maintain telomere length and continuous cell division. It is not easily envisioned how this outcome may result from signaling from other DNA regions.
In vivo, preferential damage to telomeres has been demonstrated (32). Hewitt et al have shown that telomere associated foci (TAFs) induced by x-ray irradiation and other DNA damaging agents are more numerous and remain longer than genomic DNA foci. Telomerase expression and telomere length do not influence this result. Consequently the relative proportion of TAFs increases with age. Interestingly, TAFs were frequently detected in the transit amplifying zone of intestinal crypts of old mice but not at the bottom of the crypts where intestinal stem cells are localized(32)
5.Replication Stress Associated with Developmental Blocks Underlies Cancer
Transformation
Examination of hematopoietic development showed that any developmental block at any step of the differentiation ladder is practically always associated with leukemic transformation [33]. On this basis, it was proposed that replication stress induced by blocked differentiation induced transformation by disrupting telomere integrity and/or the re-expression of telomerase [33]. Developmental blocks create favorable conditions for DNA damage by altering chromatin structure, delaying or stalling transcription, and generating recombination breaks that, when not properly handled, can be converted to single- and double-stranded DNA breaks. Moreover, it has been shown that cells unable to transition to the next differentiation step proliferate extensively, and, consequently, a local shortage of nucleotides may be generated, an important feature of replication stress [29,30].
- 6. A DNA Damage Response Is Triggered by Oncogenes
Overexpression of Myc in squamous epithelium using K5-Myc transgenic mice demonstrated that Myc induced multiple γH2AX and phospho-SMC1 foci similar to those induced by ionizing radiation (IR) as well as malignant transformation (34)
It has been reported that forced expression of other oncogenes, including E2F1, cyclin E, or cdc25A, induce the ATM signaling pathway [34].
7.Defects of chromatin remodelling factors and minor chromatin alterations other than DSB sor SSBs can also induce telomere damage and are associated with cancer
The SWI/SNF chromatin remodeling complex plays an important role in resolving R-loop conflicts. Consequently, malfunction of this complex may result in telomere dysfunction. Deregulation of SWI/SNF has been observed in undifferentiated/rhabdoid-type tumors [35,36]. This suggests that it functions at a very early stage of cell differentiation and its impairment at this stage is reflected in the very immature phenotype of the tumor cell population.
BCL11B is a transcription factor that interacts with nucleosome remodeling and the histone deacetylation complex NuRD [37]. It may also play a role in telomere structure and function [38,39]. Mutations in BCL11B have been associated with lineage ambiguous leukemias [40] and undifferentiated nervous tissue tumors [41]. There are several other cases where chromatin remodeling dysfunction is associated with acute leukemias and sarcomas with an extremely undifferentiated phenotype, suggesting that these tumors arise through errors produced in very early differentiation events at the immediate post-stem cell stage.
8.Telomere Complex Alterations Can Directly Induce Cancer
Cancer Induced by the Sheltering Protein TRF2
Whereas telomere damage appears to be an intermediate step in cancer transformation induced by all aforementioned pathways, aberrant expression of some factors from the telomere complex has also been associated with cancer transformation.
TRF2 is abundantly expressed in different cancers apart from skin carcinomas, such as breast, liver, and lung carcinomas and expression of TRF2 under the control of Keratin5 promoter was shown to lead to increased UV-induced tumors as well as spontaneous cancer formation [42,43]. When successive generations of Terc−/− mice were crossed with K5TRF2 mice to study the presumed protective role of short telomeres on tumor development, it was found that telomerase deficiency in the K5TRF2/Terc−/− mouse model led to an increased frequency of chromosome aberrations consistent with increase telomere shortening caused by TRF2, which adds to the effect of Terc deletion. However, carcinogenesis was also significantly promoted [42,43]. Interestingly reversal of telomere shortening was evident in late generations of K5TRF2/Terc−/− mice together with increased intensity of telomere fluorescence, telomere elongation, and other parameters indicative of an ALT mechanism, such as elevated numbers of telomere sister chromatide exchanges (T-SCE) and colocalization of PML with telomeres [44]
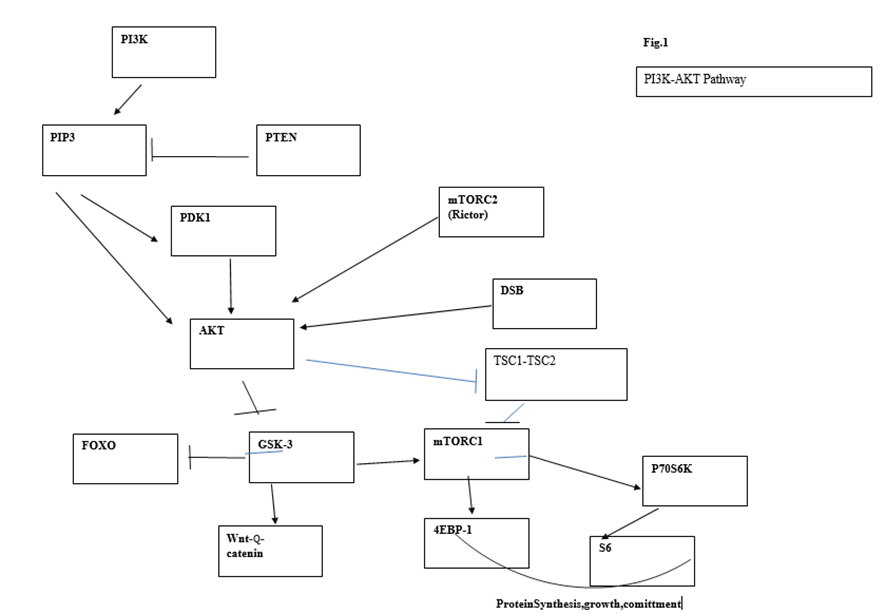
9.The PI3K/AKT pathway
Downstream targets of this pathway can mediate oncogenic effects in several directions, that is, they may induce telomere damage and connect telomere disruption to carcinogenesis as well as induce stem cell exit as a preliminary step for transformation
The PI3K/AKT pathway is connected to the DNA damage response and can be activated following DNA damage. Bozulic et al. [45] reported that Akt/Pkb is activated by one of the central kinases of the DNA damage response pathway, DNA-PK, following double-strand breaks. It has also been shown that pTen, the phosphatase that antagonizes AKT activation, is a target of ATM, the main central sensor of the DDR pathway [46]. The PI3K/AKT pathway is the most frequently mutated pathway in cancer. Some of its individual components, such as p110α and Pten, are among the most frequently mutated genes in cancer; however, many other lesions that activate this pathway are also involved in tumorigenesis (oncogenic Ras, AKT, loss of LKB1, etc.) [47].
The pathway is initiated by the conversion of phosphatidylinositol (3,4) bi-phosphate (PIP2) into the second messenger (PIP3). PIP3 binds PKB/AKT, provoking a conformation change which allows protein PDK1 to phosphorylate AKT at position T308. Full activation of AKT requires phosphorylation of AKT at Ser473 carried out by mTORC2. The phosphatase and tensin analog (Pten) negatively regulates AKT through its phosphatase activity, but Pten mutations outside the phosphatase domain demonstrate an additional role for Pten in the control of chromosome stability. Nuclear Pten binds centromeric protein C (CENP-C) and targets different proteins such as focal adhesion kinase (FAK). Pten can participate in DSB repair through induction of RAD51 and may regulate the cell cycle through the PI3K-p27-CDK2 axis [48, 49]. In agreement with the central and many-branched role played by Pten, deletion of this gene represents one of the more effective and faster means of cancer induction. Given the complexity of signaling pathways arising from this central node, numerous studies have been devoted to dissecting the impact of Pten deletion, Akt/Pkb activation, and their downstream targets in tumor development. Phosphorylation of Akt is followed by phosphorylation of GSK3β, FOXO1, and mTORC1.
Some initial studies suggested that transformation in the PI3K pathway was mediated exclusively via mTORC1 activation [50]. Partial reduction of Akt through Akt1 deletion significantly reduced skin carcinogenesis as well as Harvey Ras-induced tumors. Deletion of both Akt1 and Akt2 resulted in almost complete resistance to oncogenic transformation. In subsequent experiments, the same authors observed that Tsc2 deletion led to high mTORC1 activity and enhanced cellular replication, as measured in vitro, while Akt phosphorylation was diminished as a result of a feedback inhibitory loop. On the contrary, increased Tsc2 expression led to downregulated mTORC1 activity, decreased number of foci, and increased Akt activity. From these results, they concluded that Akt-mediated transformation relied mainly or exclusively on mTORC1 activity. However, this conclusion was based on the presumed absolute equivalence of in vitro proliferation foci with oncogenesis in vivo. The latter was not measured in these experiments. mTORC1 controls coordination of ribosome biogenesis and cell growth through its downstream targets S6 and 4E-BP1s. mTORC1 regulation of ribosome biogenesis requires S6 kinase and is mediated by upstream binding factor (UBF), a cofactor of RNA polI [51]. Suppression of UBF in the face of unaltered levels of other proliferation-associated proteins has been shown to lead to apoptosis [52, presumably due to cell proliferation unsupported by lack of protein synthesis. In addition, the inhibition of mTORC1 function by conditional deletion of Raptor, a component of the mTORC1 complex, revealed that phosphorylation of p70S6 and 4E-BP1 was dependent on the cell developmental stage. Consequently, a faulty mTORC1 function is more disruptive for transit, amplifying cells that are actively replicating and need a higher protein supply than stem and quiescent cells. This is reflected in the response of different populations of acute leukemia cells (AML) to mTORC1deprivation. mTORC1 loss leads to apoptosis of mature cells and reduction of tumor burden but does not affect leukemia stem cells (LSCs) (53)
After Pten deletion, mice develop hematological malignancies, the most prevalent of which is T-cell acute leukemia/lymphoma. The incidence of malignancies was decreased in Pten/Raptor double knockouts but incidence was still significant. In contrast, this study did not find acute leukemia/lymphoma development in Pten, Rictor double-deleted mice. Chen et al. [54] induced mTORC1 activation by conditional Tsc1 deletion. mTORC1 activation induces HSC proliferation followed by exhaustion. It was shown that increased ROS levels were responsible for HSC exhaustion because treating the mice with the antioxidant N-acetylcysteine prevented bone marrow hypocellularity and restored reconstitution capacity. However, in contrast to Pten deletion, which, like Tsc1 deletion, results in mTORC1 activation, leukemogenesis did not follow HSC exhaustion in Tsc1 KO HSCs. Contrary to the restoration of repopulating ability by N-acetylcysteine observed in Tsc1 null-HSCs, the loss of repopulation ability caused by Pten deletion could not be restored by antioxidant treatment [55]. These discrepancies might be due to the different experimental models used, but are more likely due to a greater impairment of HSC function of Pten null HSCs.
Similarly, in leukemias associated with Pten chromosomal translocations, it has been repeatedly observed that inhibition of mTORC1 signaling by rapamycin delays the onset of leukemia and reduces tumor burden but it does not affect the leukemia stem cell. From a clinical perspective, it is worth noting that mutations in TSC1 or TSC2 are responsible for tuberous sclerosis, which is associated with hamartomas rather than carcinomas. Nevertheless, mTORC1 contributes to malignant transformation induced by Akt activation because rapamycin decreases the incidence of T-cell lymphomas in murine models using constitutively active AKT [56]. Thus, the high efficiency of the PI3K/AKT/Pten pathway in promoting leukemia may result from the convergence of oncogenic signals mediated by several downstream targets of this pathway. This is consistent with the concept that cell proliferation may act in concert with deregulation of the mitotic cycle to elicit oncogenic transformation.
GSK3β is a downstream node of the PI3K pathway from which divergent signaling for HSC self-renewal and lineage commitment emanate. Activated Akt phosphorylates GSK3β, leading to its inhibition and subsequent activation of Wnt-βcatenin signaling which enhances HSC self-renewal. Concomitantly, GSKβ inhibition also activates mTORC1, promoting lineage commitment. By simultaneously stimulating the Wnt-βcatenin pathway with inhibitors of GSKβ CHIR99021 or lithium and inhibiting the mTOR pathway with rapamycin, Huang et al. [57] were able to maintain long-term multilineage hematopoiesis in cytokine free cultures treated with both inhibitors. These culture conditions allowed tri-lineage hematopoietic reconstitution and gave rise to mature myeloid cells, contrary to control cells cultured in a standard cytokine cocktail. A higher percentage of c-Kit+ quiescent cells in G0 as well as a small increase in S/G2/M, indicating some increase in cell growth, were seen in the treated cultures. Transplantation to secondary recipients confirmed the improved preservation of HSC function as assessed by a higher chimerism detected in recipients of cells treated with inhibitors [57). Although HSC proliferation and subsequent exhaustion mimic similar outcomes induced by Pten deletion, there are important differences that have been revealed by these experiments. First, HSC exhaustion induced by GSKβ inhibition is reached more slowly and becomes evident only through serial transplants and competitive repopulation assessment. More importantly, Pten loss or constitutive Akt activation leads to the rapid development of leukemia which has not been observed so far in GSK3β-rnai transplanted mice or lithium, whose use in the treatment of bipolar disorders has never been associated with increasing risk of malignancies [58]. In addition, the preservation of physiological tri-lineage hematopoietic differentiation induced by Wnt-β-catenin signaling contrasts with the biased expansion of immature myeloid progenitors subsequent to HSC mobilization observed after Pten deletion or Akt constitutive activation [56]. Again, exit from the stem cell pool appears as a step to malignant transformation. These observations suggest that cell proliferation induced by mTORC1 is not sufficient to induce transformation, and point to an essential oncogenic contribution from FOXO, another downstream target of activated AKT.
The FOXO group of transcription factors (FOXO1 (FXHR), FOXO3a (FKHRL1), and FOXO4 (AFX)) act downstream of the PI3K/AKT pathway [59] Interestingly, sites of Akt phosphorylation at threonine 308 (pAKTThr308) and Serine 473 (pAKTSer473) are essential for full Akt activation, but pAKTSer473 is dispensable for AKT-mediated phosphorylation of TSC2 and GSK-3β and is required for phosphorylation and inactivation of FOXOs [60).
FOXO proteins are normally present in an active state in the cell nucleus where they are involved in cell cycle arrest or apoptosis (depending on the physiological and cell context), control of cell cycle progression (e.g., induction of cdk inhibitor p27kip1 and p57 as well as long-term survival and control of factors involved in stemness such as OCT4 and SOX2 [59,61,]. Nuclear exclusion (inactivation) of Foxo3a is associated with the first step in hematopoietic differentiation, as Foxo3a is present in the nucleus of freshly isolated Foxo3a+/+ CD34− KSL cells (HSCs) but appears in the cytoplasm of freshly isolated Foxo3a+/+CD34+ KSL cells (progenitors) 61]. Clonogenic assays show that Foxo3a deficiency impairs the long-term (16 weeks) reconstitution ability of CD34− KSL (HSCs) but not the short-term reconstitution capacity of CD34+ KSL cells (progenitors). The decline of repopulation efficiency of HSCs is associated with loss of quiescence and decreased levels of cell cycle inhibitors p27 and p57 in Foxo3a−/− CD34− KSL cells [61]. Active FOXOs play an essential role in the maintenance of stemness (59) consequently, loss of stemness and exit from quiescence can result from Foxo nuclear exclusion and inactivation.
Finlay et al. [62] showed that “PDK1 has an obligatory function in controlling the phosphorylation and transcriptional inactivation of Foxo1, 3a and 4 in Pten-null cell” and that “Pten null T cell progenitors cannot transform or develop into invasive and fatal T lymphoma without PDK1”. Other findings lend further weight to the role of this oncogenic mechanism, such as the requirement of the mTOR complex 2 for development of prostate cancer in Pten null mice [63) or the suppression of leukemogenesis in Pten null mice by concomitant deletion of Rictor (an essential component of mTORC 2) [64] (there has been a hot debate on the role of PDK1 in Akt phosphorylation at Ser473). Other kinases referred to as PDK2 activity, and more recently mTORC2, have shown to be responsible for AktSer473 phosphorylation [65). Nevertheless, this does not invalidate the conclusions of Finlay or the ideas defended here that boil down to the paramount role of Foxo inactivation induced by pAktSer473 on T-cell lymphomagenesis [64. Magee et al. showed that Rictor deletion with the subsequent suppression of mTORC2 prevented leukemogenesis and HSC depletion in Pten-deleted adult mice [64). The expression of myr-AKT in HSCs that is associated with HSC mobilization and exit from the stem cell compartment must correlate with inactivation and cytoplasmic translocation of Foxo 3a (the main form of Foxo in hematopoietic cells) as it has been observed that transition of HSCs to progenitors is associated with inactivation and cytoplasmic translocation of Foxo [61). In normal HSCs, the activity of Akt is attenuated as required for quiescence, whereas Akt activation rises in normal granulocyte-macrophage progenitors (GMPs). Paradoxically, Sykes et al. observed growth suppression in the AML-AF9 leukemia model [66] after enforced activation of AKT in HSCs by means of the MSCV-IRES-GFP-myr-Akt construct. The leukemia stem cell (LSC) shared the immunophenotype of GMPs (lineage low, c-Kit high, FcγRII/III+, CD34+) but had the attenuated Akt pattern of a normal stem cell associated with active nuclear Foxo. Since this expression pattern seems to be related to maintenance of stemness, it is probably a general feature of the transformation process rather than a specific feature of the AML-AF9 leukemia. This suggests that the differentiation of HSCs into committed progenitors is an obligatory step in the process of transformation after which the committed progenitor must undergo a partial reversion in order to acquire the attenuated pattern of Akt expression that supports quiescence. A possibility was that activation of mTORC1 induced by pAKT with subsequent differentiation of tumor cells was responsible for this paradoxical beneficial role of pAKT in leukemic growth. Akt activates mTORC1 by relieving inhibition of mTORC1 by TSC2. As this explanation was ruled out under rapamycin treatment, other actions of pAkt, independent of mTORC1 activation, must be responsible for myeloid maturation and growth inhibition. Given the substrate selectivity of pAKTSer473, the involvement of Foxo inactivation was an obvious choice. This was confirmed by inhibiting Foxo3a with shRNA in MLL-AF9 as well as in AML cell lines that do not carry MLL translocations [66). Foxo inhibition lowered tumor growth and induced myeloid-maturation-related death, but Foxo inhibition does not affect the generation or survival of leukemic stem cells. Thus, pAKTSer473 induced Foxo3a cytoplasmic translocation and exit from the HSC compartment (commitment) concomitantly, but generation of the leukemia stem cell involves reversion of a committed cell to the attenuated AKT pattern of the normal stem cell. In other words, HSC differentiation and subsequent leukemic transformation are two outcomes of Foxo3a signaling that affect tumor biology in opposing ways, but once leukemic transformation is established only Foxo’s deactivation role on cell differentiation is observed. Obviously, deletion of Foxo in AML-F9 transplanted Cre+ recipient mice extended latency and survival due to differentiation-related cell death, but the majority of mice eventually succumbed to leukemia, suggesting that, independently of its effect on myeloid maturation and apoptosis, Foxo3 depletion cannot eradicate LSCs after transformation. Collectively, these results imply that putative transformation of a stem cell requires a previous step of differentiation (Figure 1).
Hu et al. [67 revealed an Akt-independent mechanism of Foxo3a inactivation dependent on IκB kinase (IKK). Constitutive expression of IκB kinase leads to Foxo3a inactivation and nuclear exclusion, cell proliferation, and tumorigenesis, a clear indication that the proleukemogenic role of Foxo3a predominates over its antileukemic effect. Degradation of IκBα by IκB kinase is accompanied by the activation and nuclear translocation of NF-κB, which is known to be associated with the upregulation of cyclin D1 and cell proliferation. However, tumorigenesis was attributed to Foxo inactivation because cell clones transfected with, and expressing, IκB kinase induced mammary tumors in nude mice which could be suppressed by re-expression of Foxo3a [67) . The interplay between Foxo and NF-κB may be crucial for the integrity of the cancer stem cell and this interplay may be influenced by telomere alterations through the demonstrated ability of telomere protein Rap1 to bind to IKK, thereby promoting degradation of IκB and subsequent translocation of NF-κB to the nucleus [68]. The same outcome (IKK activation and NF-κB nuclear translocation) can be elicited by DNA double-strand breaks (DSBs) but not by proinflammatory stimuli [69). Following DSBs, ATM can activate IKK, leading to the degradation of IκB and release of bound NF-κB which translocates to the nucleus. In contrast to upregulation of cyclin D1 expression, antiapoptosis, and cell proliferation associated with nuclear NF-κB, nuclear FOXO drives cell cycle arrest or apoptosis, whereas cytoplasmic Foxo drives cell proliferation. Under physiological conditions, AKT activity is low in quiescent cells and it is associated with retention of FOXO factors in the nucleus, where they upregulate expression of target genes that control cell cycle, i.e., p27kip1, Rb2(p130), mitosis (cyclin B and polo-like kinase, metabolism, or apoptosis (Fas ligand and Bim)) [59,60,65,67 70). Some of these phenotypic traits, like upregulation of p27 and cell cycle arrest, can also be induced by re-expression of Pten. In contrast, Pten deletion and subsequent Akt activation led to FOXO inactivation and nuclear exclusion, and presumably cycle control deregulation. The proapoptotic function of Foxo proteins is in great part dependent on their collaboration with E2F1 protein (70) This collaboration is required for E2F1-induced apoptosis but not for E2F1-induced proliferation. A protective DNA damage reaction increases apoptosis through E2F1-Foxo because it stabilizes E2F1 through ATM and Chk2 phosphorylation, whereas the Foxo-dependent arm of E2F1 signaling in charge of apoptosis was found to be reduced in most tumor types examined relative to counterpart normal samples. These findings highlight the relevance of the cross-talk between the oncogene-induced DNA damage reaction and the PI3K pathway [70]. Pin1, a target of E2F1 essential for Neu/Ras-induced transformation of mammary epithelial cells through activation of cyclin D1 [71), has also been reported to induce nuclear accumulation of NF-κB.
The simultaneous cytoplasmic translocation–inactivation of Foxo leading to deficient apoptosis, differentiation (exit from the stem cell pool) and prosurvival effect, and induction of cell proliferation by nuclear translocation of NF-κB appear to reinforce each other in the transformation and survival of the cancer stem cell.
Figure 1. Main circuits of the PI3K/AKT pathway and its connection to DSBs.
10.Resistance of Neonatal Cells to Leukemia Development
The Ser473 AKT mTORC2 site required to inactivate Foxo3a is also required to mobilize HSCs, as demonstrated by the decrease in the S/G2/M fraction of HSCs after Pten deletion when Rictor, a component of mTORC2, is deleted. The mobilization of HSCs has an important proleukemogenic effect, as demonstrated by the antileukemic role of Rictor deletion in Pten-deficient cells (72]. Rictor deletion, but not rapamycin treatment, reduced HSC mobilization after Pten deletion. Rapamicin treatment and Rictor deletion both reduced HSC proliferation after Pten deletion and additively reduced the severity of myeloproliferative disorder following Pten deletion.
An experiment by Xue et al. [72) revealed a special form of cancer resistance linked to cell or organ immaturity, which may underlie the resistance of stem cells to cancer development. They described a different behavior of HSC in neonatal mice compared to that in adult mice in their response to Pten deletion. When Pten deletion was performed 2 days after birth, the expected increase in HSCs number was only observed in 8-week-old mice (>40-fold) but it had no effect on the number of HSCs in 2-week-old mice. Overall proliferation of unfractionated bone marrow cells at 2, 3, 4, and 5 weeks of age was not affected by Pten deletion. On the other hand, control neonatal HSCs divided more actively than adult HSCs, with transition from neonatal to adult phenotype taking place between 4–5 weeks. In transplantation experiments, Pten-deficient neonatal (before 6 weeks of age) HSCs showed long-term multilineage reconstitution lasting from 16 to 24 weeks, and even this constraint (16–24 weeks) appears to reflect their maturation to an adult phenotype. In contrast, Pten-deficient adult HSCs showed only short-term reconstitution lasting less than 16 weeks. It was also found that Pten prevented leukemia of adult but not neonatal mice. The fact that adult mice succumbed to leukemia 12 weeks after Pten deletion made it difficult to test whether neonatal mice were resistant to leukemia, since their HSCs would mature before the onset of leukemia. To circumvent this difficulty, pIpC was administered to Mx1-Cre, Ptenfl/fl, p53−/− mice, and littermate controls lacking Mx-1-Cre at 2 days or 6 weeks after birth. All mice in the 6 weeks group died before 20 days of pI-pC treatment, whereas at 21 days after pI-pC treatment, none of the doubly deficient Pten, p53 neonatal mice showed any sign of illness and did not die until 44 to 60 days after pI-pC treatment. Additionally, comparison of PI3K kinase activation in Pten-deleted neonatal and adult mice showed that Pten deletion increased phosphorylation of most substrates of the pathway (AKT, S6, and GSK3β, but not MAPK or AMPK) in LSK from adults but not neonatal mice. Nevertheless, HSCs from neonatal mice can activate this pathway, as demonstrated in in vitro cultures with 2% bovine serum. AKT phosphorylation at Ser473 distinguished Pten-deficient HSCs from HSCs that are dividing under physiological conditions, and Rictor deletion, which suppresses mTORC2 function, substantially reduced AKTSer473 phosphorylation, consistent with mTORC2 being less active in neonatal HSCs. Rapamicin did not affect AKTSer473 but Rictor deletion modestly reduced mTORC1 (modest reduction in S6 phosphorylation). Proliferation of HSCs after Pten deletion depends on mTORC1 as it is reduced by rapamycin. In contrast, Rictor deletion in Pten null HSCs reduced the frequency of HSCs in cycle. Also, in adult mice, Rictor deletion significantly reduced MPPs but not other lineages. All these findings show that mTORC1 induces HSC proliferation–differentiation and is the main regulator of these activities under physiological conditions, whereas Pten deletion induces HSC proliferation and mobilization of HSCs through mTORC2-dependent and -independent mechanisms that do not mimic physiological HSC self-renewal. These findings are consistent with the role previously described of AktSer473 in relation with Foxo. Moreover, Rictor deficiency in Pten null cells restored long-term reconstitution ability (of myeloid and T cells, although not B lymphoid cells), which suggests that loss of long-term reconstitution is associated with HSC mobilization (and accelerated differentiation to MPPs since Rictor deletion reduced MPP number) and, in turn, prevention of leukemia development by Rictor deletion correlates with restored repopulation efficiency due to decreased HSC mobilization and early differentiation to MPPs.
Therefore, the resistance of neonatal cells to leukemia development appears to depend on a deficient mTORC2 activity in neonatal HSCs. However, the question remains whether antileukemic protection afforded by deficient mTORC2 activity may do so by preventing exit from the stem cell pool and loss of self-renewal potential associated with stem cells.
In addition, the work of Xue et al. [72) demonstrates the role of a DNA damage response in the initiation of oncogenesis through the PI3K/AKT pathway. T-cell thymomas that develop in Pten-deficient mice are CD4+ CD8− mature T-cell thymomas. These researchers observed that premalignancy only starts at 9 weeks of age in the preceding stage of double-positive (DP) thymocytes and is accompanied by classical markers of a DNA damage reaction such as phosphorylation of DDR substrates p53, chk1, and chk2 in DP thymocytes. These markers were highly phosphorylated in w.t. thymocytes consistent with the stage where V(D)J recombination occurs (it was negligible in CD4SP and naïve w.t. T cells). Pten-deficient DP thymocytes expressed higher levels of these proteins but not of γH2AX (no thymocyte populations expressed γH2AX). They also expressed higher levels of p19 and p21. AKT phosphorylation was not detected before 6 weeks of age in Pten-deficient T cells. Concomitantly, Foxo3a was heavily phosphorylated in DP T cells but not in SP or naïve T cells of Pten-deficient 9-week-old mice. GSK3β was only slightly activated but S6K was unaffected. They concluded that, although Pten is lost since birth, premalignancy does not start until mice are 6 weeks old and activation of AKT in DP cells differentially affect downstream targets of the PI3K pathway. DP thymocytes from both controls and Pten-deficient cells were Ki67 positive (a marker of non-G0 cells) but did not proliferate. The most striking changes were seen in the expression of p27 that was expressed at high level in w.t. DP thymocytes and was significantly reduced in Pten-deficient DP thymocytes [72].
The association of stem cell resistance to leukemogenesis with the need for a short period of organismal maturation before a cell becomes vulnerable to cancer transformation points to obscure mechanism/s of anticancer protection linked to the cell immaturity, which may underlie the observation that, contrary to a widespread assumption, there are few true stem cell cancers. On the other hand, the synchronous appearance of premalignancy at the DP stage suggests that the DNA strand breaks that occur at this developmental stage along with V(D)J recombination initiate a DDR that, in conjunction with Pten deficiency, is responsible for malignant transformation.
11.Further Comments and Conclusions
A faulty repaired DNA may lead to mutated DNA, leading to enhanced cell proliferation or, if unrepaired, to cell death, but it would be the subsequent checkpoint defect perhaps boosted by stimulation of cell proliferation that may induce cell cycle deregulation and telomere dysfunction ultimately giving rise to the emergence of a new mechanism of telomere maintenance. Furthermore, even in the absence of direct disturbance of the cell cycle, DNA alterations may directly impact the telomere. For instance, it has been reported that deficiency of DNA mismatch repair increases the rate of telomere shortening [73].
Evidently, DDR occurs more frequently during tissue turnover and differentiation than in the stem cell compartment leading to disturbances of the telomere complex (erosion, ALT, gain-of-function mutations at the TERT promoter) or telomerase re-expression (or one of its downstream targets), which has been demonstrated to induce transformation independently of its canonical enzymatic activity. telomerase through its downstream target TGFβ and TGFβ target JunB [16,74)
However, there are rare cases of telomerase oncogenic pathways starting directly in stem cells (primary defects of the telomere complex). One such case is represented by Beckwith–Wiedemann syndrome, which is associated with an 800-fold increased risk of childhood neoplasms, where multiple tumors arise in different organs or even in the same organ. Overexpression of telomerase reverse transcriptase (TERT), defective TGFβ signaling with epigenetic silencing of β2 spectrin, an SMAD adaptor for TGFβ signaling, are involved in the etiology of this syndrome [13]. Another examples of this, in the area of molecular cancer modeling, were the reports by Passegué et al. [14] and Santaguida et al (15] that JunB inactivation recapitulates aspects of human malignancies, including myelogenous leukemia.
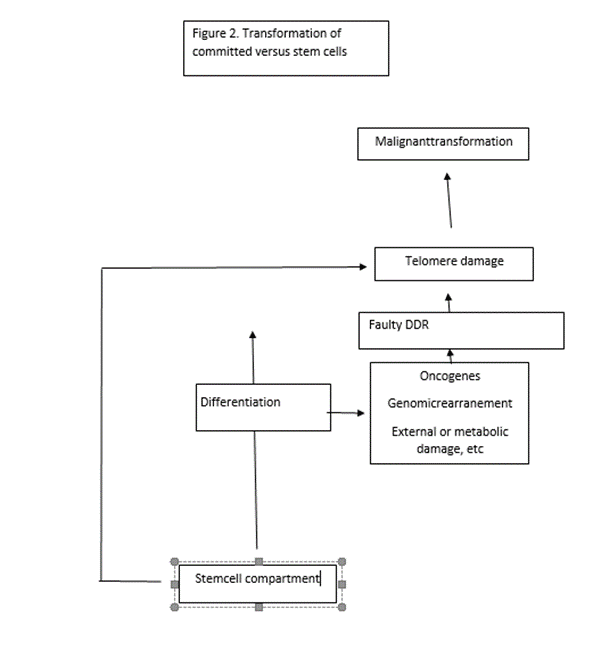
According to the above, there are two main oncogenic routes: one initially triggered by DNA damage reactions that converge on telomere alterations ultimately responsible for oncogenic signaling (this mechanism would be preferentially associated with tumors originating in relatively mature cells); and another one that is initiated by mechanism directly involved in telomere maintenance represented by tumors of true stem cell origin. (Fig.2) However, this may be a simplification because there are oncogenes that may potentially act through both routes. For instance, overexpression of Tcl1 has been shown to cause B-CLL by inhibiting AP-1 (containing JunB) and enhancing NF-κB [75] Fanconi anemia is a recessive genetic disorder that manifest early in life and evolves with bone marrow failure due to decrease of hematopoietic stem cells which is frequently followed by acute leykemia. These clinicopathological features meet the criteria set above for inclusion in the group of childhood/immature cancers and as the above discusión indicates is a component of the DDR pathway that may directly cause telomere dysfunction.
REFERENCES
- Torres-Montaner, A & Hugues D. A hypothetical anti-neoplastic mechanism associated to reserve cells. J. Theoretical Biolgy (2004), 231, 239-248
- Cancer origin in committed versus stem cells: Hypothetical antineoplastic mechanisms associated with stem cells. Critical Rev. Oncol./Hematol. 2011, 80, 209–224.
- Chin, K.; Ortiz de Solorzano, C.; Knowles, D.; Jones, A.; et al. In situ analysis of genome instability in breast cancer. Genet. 2004, 36, 984–988
- Ventura-Ferreira, M.S.; Crysandt, M.; Ziegler, P.; Hummel, S.; et al. Evidence for a pre-existing telomere deficit in non-clonal hematopoietic stem cells in patients with acute leukemia. Hematol. 2017, 96, 1457–1461.
- Wilson, A.; Murphy, M.J.; Oskarsson, T.; Kaloulis, K.; et al. c-Myc controls the balance between stem cell self-renewal and differentiation. Dev. 2004, 18, 2747–2763.
- Wang, J.; Xie, L.Y.; Allan, S.; Beach, D.; et al. Myc activates telomerase. Genes Dev. 1998, 12, 1769–1774.
- Nepal RM, Tong Li, Kolaj B, Edelmann W and A. Marin. Msh2-dependent DNA repair mitigates a unique susceptibility of B cell progenitors to c-Myc-induced lymphomas.PNAS 106(44): 18698-18703).
- Liu G, Elf SE, Miyata Y, Sashida G. et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell (2009), 4, 37-48
- Aasai T, Liu Y, Bae N, Nimer SD. The p53 tumor suppressor protein regulates hematopoietic stem cell fate. J. Cell Physiol (2011), 226(9), 2215-2221
- Liu G, Parant JM, Lang G, ChauP, Chavez-Reyes A et al Chromosome stability in the absence of apoptosis is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat Genet (2004), 36(1), 63-68
- Prokocimer M, Molchadsky A, Rotter V. Perspective dysfunctional diversity of p53 proteins in adult acute myeloid leukemia: projections on diagnostic workup and therapy. Blood (2017) 130, 699-712
- Yamanaka, S. Pluripotent stem cell-based cell therapy-promise and challenges. Cell Stem Cell 2020, 27, 23–531.
- Chen, J.; Tsukamoto, H.; Mushra, L.; Chen, J.; et al. Suppression in human stem cell disorder Beckwith-Wiedemann syndrome find the latest version: TGF-β/β2-spectrin/CTF-regulated tumor suppression in human stem cell disorder Beckwith-Wiedemann syndrome. Clin. Investig. 2016, 126, 527–542.
- Passegué, E.; Wagner, E.F.; Weissman, I.L. JunB deficiency leads to a myeloproliferative disorder arising from hematopoietic stem cells. Cell 2004, 119, 431–443.
- Santaguida, M.; Schepers, K.; King BSabnis, A.J.; et al. JunB protects against myeloid malignancies by limiting hematopoietic stem cell proliferation and differentiation without affecting self-renewal. Cancer Cell 2009, 15, 341–352.
- Geserick C, Tejera A, Gonzalez-Suarez E, Klatt P. et al Expression of mTert in primary murine cells links the growth-promoting effects of telomerase to transforming growth factor-β-signaling. Oncogene (2006), 25, 4310-4319
- Torres-Montaner A. Interactions between the DNA damage response and the telomere complex in carcinogénesis: A Hypothesis. Current issues in Molecular Biology (2023), 45, 7582-7616. https://doi.org/10.3390/cimb45090478
- Tanaka, A.; Weinel, S.; Nagy, N.; et al. Germline mutation in ATR in autosomal dominant oropharyngeal cancer syndrome. Am. J. Hum. Genet. 2012, 90, 511–517.
- Flynn, R.L.; Zou, L. ATR: A master conductor of cellular responses to DNA replication stress. Trends Biochem. 2011, 36, 133–140.
- Ruzankina, Y.; Pinzon-Guzman, C.; Asare, A.; et al. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell 2007, 1, 113–126. https://doi.org/10.1016/j.stem.2007.03.002.
- Epanchintsev, A.; Costanzo, F.; Rauschendorf, M.-A.; et al. Cockayne’s syndrome A and B proteins regulate transcription arrest after genotoxic stress by promoting ATF3 degradation. Cell 2017, 68, 1054–1066. https://doi.org/10.1016/j.molcel.2017.11.009.
- Iyama, T.; Wilson, D. III. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair 2013, 12, 620–636. https://doi.org/10.1016/j.dnarep.2013.04.015
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; et al. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003, 22, 5612–5621.
- Matei, I.R.; Guidos, C.J.; Danska, J.S. ATM-dependent DNA damage surveillance in T-cell development
- Tauchi, H.; Matsuura, S.; Kobayashi, J.; Sakamoto, S.; Komatsu, K. Nijmegen breakage syndrome gene, NSB1, and molecular links to factors for genome stability. Oncogene 2002, 21, 8967–8980.
- Attwooll, C.L.; Akpmar, M.; Petrini, J.H.J. The Mre11 complex and the response to dysfunctional telomeres. Cell. Biol. 2009, 29, 5540–5551.
- Spehalski, E.; Capper, K.M.; Smith, C.J.; Morgan, M.J.; et al. MRE11 promotes tumorigenesis by facilitating resistance to oncogene-induced replication stress. Cancer Res. 2017, 77, 5327–5338. https://doi.org/10.1158/0008-5472CAN-17-1355
- Liao M-J, Zhang X-X, Hill R, Gao J, Qumsiyeh MB, Nichols W, van Dyke T. No requirement for V(D)J recombination in p53-deficient thymic lymphoma. Mol Cell Biol 18(6), 3495-3501
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011, 145, 435–446.
- Saldivar, J.C.; Miuma, S.; Bene, J.; Hossein, S.A.; Dhibata, H.; et al. Initiation of genome instability and preneoplastic processes through loss of Fhit expression. PLoS Genet. 2012, 8, e1003077.
- Hemanth Tummala, Amanda Walne, Roberto Buccafusca, Jenna Alnajar, Anita Szabo et al. Germline thymidylate synthase deficiency impacts nucleotide metabolism and causes dyskeratosis congenita The American Journal of Human Genetics (2022) 109, 1472– 1483 http://doi.org/10.1016/j.ajhg.2022.06.014
- Hewitt G, Jurk D, Marques FDM, Correia-Melo C et al. Nature communications (2012) 3, 708- doi: 10.1038/ncomms1708
- Torres-Montaner. The telomere complex and the origin of the cáncer stem cell. Res. 2021, 9, 1–30. https://doi.org/10.1186/s40364-021-00339-z
- Pusapati, R.V.; Rounbehler, R.J.; Hong, S.; Powers, J.T.; Yan, M.; et al. ATM promotes apoptosis and suppresses tumorigenesis in response to Myc. Natl. Acad. Sci. USA 2006, 103, 1446–1451.
- Bayona-Feliu, A.; Barroso, S.; Muñoz, S. The SWI/SNF chromatin remodeling complex helps resolve R-loop-mediated transcription-replication conflicts. Genet. 2021, 53, 1050–1063. https://doi.org/10.1038/s41588-021-00867-2.
- Schaefer, I.-M.; Hornick, J.L. SWI/SNF complex-deficient soft tissue neoplasms: An update. Diagn. Pathol. 2021, 38, 222–231.
- Goos, J.A.C.; Vogel, W.K.; Micochova, H.; et al. A de novo substitution in Bcl11b leads to loss of interaction with transcriptional complexes and craniosynostosis. Mol. Genet. 2019, 28, 2501–2513. https://doi.org/10.1093/hmg/ddz072.
- Yang, S.F.; Sun, A.A.; Shi, Y.; et al. Structural and functional characterization of RBBP4-ZNF827 interaction and its role in NuRD recruitment to telomeres. Biochem. 2018, 475, 2667–2679. https://doi.org/10.1042/BCJ20180310.
- Conomos, D.; Reddel, R.R.; Picket, H.A. NuRD-ZNF827 recruitment to telomeres creates a molecular scaffold for homologous recombination. Struct. Mol. Biol. 2014, 21, 760–770. https://doi.org/10.1038/nsmb.2877.
- Montefiori, L.E.; Mulligan, C.G. Redefining the biological basis of lineage-ambiguous leukemia through genomics: BCL11B deregulation in acute leukemias of ambiguous lineage. Best Pract. Res. Clin. Haematol. 2021, 34, 101329. https://doi.org/10.1016/j.beha.2021.101329.
- Lennon, M.J.; Jones, S.P.; Lovelace, M.D.; et al. Bcl11b-A critical neurodevelopmental transcription factor- Roles in health and disease. Neurosci. 2017, 11, 89. https://doi.org/10.3389/fncel. 2017.00089
- Blanco, R.; Muñoz, P.; Flores, J.M.; Klatt, P.; Blasco, M.A. Telomerase abrogation dramatically accelerates TRF2-induced epithelial carcinogenesis. Genes Dev. 2007, 21, 206–220.
- Muñoz R, Blasco, M.A. Blasco Role of the TRF2 telomeric protein in cancer and aging. Cell Cycle 2006, 5, 718–721.
- Imran, S.A.M.; Yazid, M.D.; Cui, W.; Lokanathan, Y. The intra and extratelomeric role of TRF2 in the DNA damage response. J. Mol. Sci. 2021, 22, 9900. https://doi.org/10.3390/ijms22189900
- Bozulic, L.; Surucu, B.; Hynx, D.; Hemmings, B.A. PKBα/Akt1 acts downstream of DNA-PK in the DNA double strand break reponse and promotes survival. Cell 2008, 30, 203–213.
- CBassi; Fortin, J.; Dnow, B.E.; Wakeham, A.; et al. The PTEN and ATM axis controls the G1/S cell cycle checkpoint and tumorigenesis in HER2-positive breast cancer. Cell Deth Differ. 2021, 28, 3036–3051. https://doi.org/10.1038/s41418-021-00799-8.
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Rev. Drug Discov. 2014, 13, 140–156. https://doi.org/10.1038/nrd4204.
- Shen, W.H.; Balajee, A.S.; Wang, J.; Wu, H. et al. Essential role for nuclear Pten in maintaining chromosomal integrity. Cell 2007, 128, 157–170.
- Planchon, S.M.; Waite, K.A.; Eng, C. The nuclear affairs of Ptem. Cell Sci. 2008, 121, 249–253.
- Skeen, J.E.; Bhaskar, P.T.; Chen, C.-C.; Chen, W.C.; et al. AKT deficiency impairs normal cell proliferation and suppresses oncogenesis in a p53-independent and TORC1-dependent manner. Cancer Cell 2006, 10, 269–280.
- Hannan, K.M.; Brandengurger, Y.; Jenkins, A.; Sharkey, K.; et al. mTOR-dependent regulation of ribosomal gene transcription requires S6K1 and is mediated by phosphorylation of the carboxy-terminal activation domain of the nucleolar transcription factor UBF. Cell. Biol. 2003, 23, 8862–8877.
- Torres-Montaner, A.; Bolivar, J.; Astola, A.; Gimenez-Mas, J.A.; Brieva JA and Valdivia MM. Immunohistochemical detection of ribosomal transcription factor UBF and AgNOR staining identify apoptotic events in neoplastic cells od Hodgkin`s disease and other lymphoid cells. Histochem. Cytochem. 2000, 48, 1521–1530.
- (Hoshii T, Tadokoro Y, Naka K, OOshio T et al. mTORC1 is essential for leukemia propagation but not stem cell self-renewal. Journal of clinical investigation (2012) 122(6), 2114-2129
- Chen, C.; et al. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. Exp. Med. 2008, 205, 2397–2408.
- Lee, J.Y.; Nakada, D.; Yilmaz, O.H.; Tothova, Z.; Joseph, N.M.; Lim, M.S.; Gilliland, D.G.; Morrison, S.J. mTOR activation induces tumor suppressors that inhibit leukemogenesis and deplete hematopoietic stem cells after Pten deletion. Cell Stem Cell 2010, 7, 593–605.
- Kharas, M.G.; Okabe, R.; Ganis, J.J.; Gozo, M. et al. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood 2010, 115, 1406–1415.
- Huang, J.; Nguyen-McCarty, M.; Hexner, E.O.; Danet-Desnoyers, G.; et al. Maintenance of hematopoietic stem cells through regulation of Wnt and mTOR pathways. Med. 2012, 18, 1778–1785.
- Huang, J.; Zhang, Y.; Bersenev, A.; O’Brien, W.T.; et al. Pivotal role for glycogen synthase kinase-3 in hematopoietic stem cell homeostasis in mice. J. Investig. 2009, 119, 3519–3529.
- Murtaza, G.; Khan, A.K.; Rashid, R.; Muneer, S.; Hasan, S.M.F.; Chen, J. Foxo transciptional factors and long-term living. Oxidative Med. Cell. Longev. 2017, 2017, 1–8. https://doi.org/10.1155/2017/3494289.
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; et al. Ablation in mice of the mTORC components raptor, rictor or mLST8 reveals that mTORC2 is required for signaling to AKT_FOXO and PKC alpha but not S6K1. Dev. Cell 2006, 11, 859–871. https://doi.org/10.1016/j.devcel2006.10.007.
- Miyamoto, K.; Araki, K.Y.; Naka, K.; Arai, F.; et al. Foxo3a is essential for the maintenance of the hematopoietic stem cell pool. Cell Stem Cell 2007, 1, 101–112.
- Finlay, D.K.; Sinclair, L.V.; Feijoo, C.; Waugh, C.M.; et al. Phosphoinositide-dependent kinase 1 controls migration and malignant transformation but not cell growth and proliferation in Pten-null lymphocytes. Exp. Med. 2009, 206, 2441–2454.
- Guertin, D.A.; Stevens, D.M.; Saitoh, M.; Kinkel, S.; Crosby, K.; Sheen, J.-H.; Mullholland, D.J.; Magnuson, M.A.; Wu, H.; Sabatini, D.M. The mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell 2009, 15, 148–159.
- Magee, J.A.; Ikenoue, T.; Nakada, D.; Lee, J.Y.; et al. Temporal changes in Pten and mTORC2 regulation of hematopoietic stem cell self-renewal and leukemia suppression. Cell Stem Cell 2012, 11, 415–428.
- Duronio, V. The life of a cell: Apoptosis regulation by the PI3K/PKB pathway. J. 2008, 415, 333–344.
- Sykes, S.M.; Lane, S.W.; Bullinger, N.; Kalaitzidis, D.; et al. AKT/FOXO signaling enforces reversible differentiation blockade in myeloid leukemia. Cell 2011, 146, 697–70.
- Hu, M.C.T.; Lee, D.-F.; Xia, W.; Golfman, L.S.; et al. IκB kinase promotes tumorigenesis through inhibition of Forkhead FOXO3a. Cell 2004, 117, 225–237.
- Cai, Y.; Kandula, V.; Ye, X.; Irwin, M.G.; Xia, Z. Decoding telomere protein Rap1: Its telomeric and nontelomeric functions and potential implications in diabetic cardiomyopathy. Cell Cycle 2017, 16, 1765–1773.
- Li, N.; Banin, S.; Ouyang, H.; Li, G.C.; et al. ATM is required for IκB kinase (IKK) activation in response to DNA double strand breaks. Biol. Chem. 2001, 270, 8898–8903.
- Shats, I.; Gatza, M.L.; Liu, B.; Angus, S.P.; You, L.; Nevins, J.R. FOXO transcription factors control E2F1 transcriptional specificity and apoptotic function. Cancer Res. 2013, 73, 6056–6067.
- Ryo, A.; Liou, Y.-C.; Wulf, G.; Nakamura, M.; et al. Pin1 is an E2F target gene essential for Neu/Ras-induced transformation of mammary epithelial cells. Cell. Biol. 2002, 22, 5281–5295.
- Xue L, Nolla H, Suzuki A, Mak TWand Winoto. Normal development is an integral parto f tumorigenesis in T-cell specific PTEN-deficient mice. PNAS (2008), 105(6), 2022-2027
- Mendez-Bermudez, A.; Royle, N.J. Deficiency in DNA mismatch repair increases the rate of telomere shortening in normal human cells. Mutat. 2011, 32, 939–946. https://doi.org/10.1002/humu.21522.
- Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, et al. Ormal and neoplastic non stem cells can spontaneously convert to a stem-like state. PNAS (2011), 108, 7950-7955
- Pekarsky, Y.; Palamarchuk, A.; Maximov, V.; Efanov, E.; et al. Tcl1 functions as a transcriptional regulator and is directly involved in the pathogenesis of CLL. Natl. Acad. Sci. USA 2008, 105, 19643–19648
(Figure 2) Different routes to malignant transformation are followed by normal stem and committed cells
This entry is adapted from the peer-reviewed paper 10.3390/cimb45090478