Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Acute respiratory distress syndrome (ARDS) is a major cause of hypoxemic respiratory failure in adults, leading to the requirement for mechanical ventilation and poorer outcomes. Dysregulated surfactant metabolism and function are characteristic of ARDS. A combination of alveolar epithelial damage leading to altered surfactant synthesis, secretion, and breakdown with increased functional inhibition from overt alveolar inflammation contributes to the clinical features of poor alveolar compliance and alveolar collapse.
- ARDS
- surfactant
- phospholipids
1. Introduction
1.1. Pulmonary Surfactant
Pulmonary surfactant is a complex mixture of phospholipids (80%), proteins (10%) and neutral lipids (10%). It is synthesized, secreted, and recycled by alveolar type-II epithelial cells (AT-II) with a primary function to reduce alveolar surface tension at the air–liquid interface, providing mechanical stability for gas exchange [1]. Surfactant components are also involved in innate immunity and are essential for the host’s defense mechanisms against infections [2][3]. The phospholipids account for most of surfactant composition with phosphatidylcholine (PC) and phosphatidylglycerol (PG) being the most abundant phospholipids. Minor phospholipids, including phosphatidylinositol (PI), phosphatidylethanolamine (PE), phosphatidylserine (PS), sphingomyelin (SM) and lysophosphatidylcholine (LPC) make up the rest of the phospholipid distribution [4][5]. The principal surface-active molecule is disaturated dipalmitoyl-PC or DPPC (PC16:0/16:0 or PC 32:0), which accounts for approximately 50% of PC [6]. Low surface tension is essential at the alveolar surface to minimize pressure gradients across the alveolar lining preventing premature airway collapse. Under dynamic compression, DPPC can reduce surface tension to near zero values in vitro [7].
There are four surfactant-based proteins, SP-A, SP-B, SP-C and SP-D. SP-B and SP-C are hydrophobic proteins involved in the adsorption of surfactant film, whereas SP-A and SP-D are hydrophilic and participate in innate immunity. Hereditary SP-B deficiency leads to lethal respiratory failure, whereas hereditary SP-C deficiency leads to acute and chronic lung diseases [8][9]. Animal models of SP-A deficiency can lead to increased susceptibility to respiratory tract infections, whereas SP-D knockout mice models demonstrate increased alveolar infiltration of macrophages, AT-II cell hyperplasia and excess phospholipid production leading to the development of emphysema [10]. Primary surfactant deficiency due to lung immaturity is the characteristic feature of neonatal respiratory distress syndrome (nRDS), where exogenous surfactant replacement is associated with improved clinical outcomes [11]. Detailed surfactant composition, metabolism and function are evaluated by excellent in-depth reviews [1][2][7][8][9][10][12][13].
1.2. Acute Respiratory Distress Syndrome (ARDS)
ARDS is a heterogenous disease process characterized by pathological changes of diffuse alveolar damage with alveolar epithelial and endothelial injury, leading to alveolar capillary leak and pulmonary oedema [14]. Clinically, patients present with poor alveolar compliance, non-hydrostatic pulmonary oedema, and hypoxemic respiratory failure [15]. It is an under-recognized syndrome even in an intensive care unit setting due to the complexity around the multi-component nature of its diagnostic definition, which has been evolving over the past 50 years [16]. According to the current Berlin definition, ARDS is diagnosed when there is an acute onset (<7 days) of symptoms, the presence of bilateral radiological opacities, varying severity of arterial hypoxemia (PaO2/FiO2 ratio < 100 mmHg: Severe, PaO2/FiO2 ratio: 100–200 mmHg as moderate, PaO2/FiO2: 200–300 mmHg as mild) and the absence of cardiogenic cause for pulmonary oedema [16].
The severity of the hypoxemia correlates with adverse outcome, where severe ARDS is associated with 40–50% mortality and the milder version with <30% mortality [16]. Sepsis from both direct and indirect lung injury is the primary risk factor for development of ARDS [17]. The mortality outcome is also variable between patients depending on the cause of ARDS. For instance, direct ARDS from pulmonary etiology associated with sepsis has a much higher mortality than-non-pulmonary ARDS without sepsis [18]. Moreover, patients with trauma related ARDS have a better prognosis than ARDS patients associated with cirrhosis or liver failure [19][20][21]. Treatment response is also vastly different between patients depending on their specific risk factors. Despite attempts to harmonize the ARDS diagnostic criteria to facilitate clinical trials, clinical heterogeneity remains a major issue [22]. Even in ARDS related to pulmonary etiology, there are variations in clinical outcomes and response from different insults such as viral infections, bacterial infections and chemical pneumonitis following aspiration of gastric contents. Recently, post hoc analyses of published ARDS randomized controlled trials suggest the existence of phenotypes according to the degree of lung and systemic inflammation as hyper-inflammatory and hypo-inflammatory with variations in responses to treatment [23].
2. Surfactant Abnormalities in ARDS
Surfactant compositional and functional abnormalities in ARDS are likely due to several reasons. The pathophysiological processes leading up to ARDS are complex and involve alveolar epithelial cellular apoptosis with significant neutrophil-mediated inflammatory infiltration, pulmonary oedema, and invasion of alveolar space by plasma and inflammatory constituents. Earlier studies on ARDS patients identified impaired surface film compressibility from lavage fluid from ARDS patients [24]. Although surfactant abnormalities are consistently seen in ARDS patients, whether they are the primary cause of lung injury, or a consequence of the initial insult is not fully understood. Several studies have characterized surfactant molecular composition and alterations in ARDS patients, which are detailed below. A summary of surfactant alterations and pathophysiological consequences in ARDS are schematically presented in Figure 1.
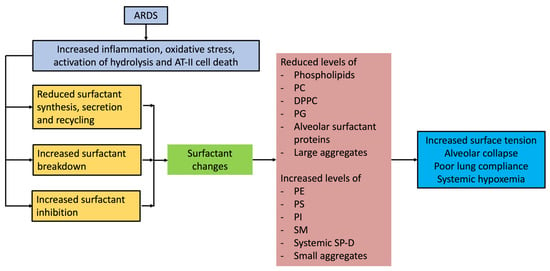
Figure 1. Surfactant alterations and the pathophysiological consequences in ARDS.
2.1. Lung Fluid Phospholipid Alterations in ARDS
ARDS patients exhibit qualitative and quantitative changes in the phospholipid composition of lung fluid recovered by bronchoalveolar lavage or tracheal aspiration. Although the total phospholipid content is variable among studies, the measurements of absolute phospholipid concentrations are limited by variability in sample recovery and analytical methods. The most common finding of surfactant alteration is related to the molecular compositional variations in phospholipid distribution. The first comprehensive surfactant phospholipid analysis in human ARDS, Hallman et al. in 1982, demonstrated low levels of lecithin (phosphatidylcholine), particularly the disaturated lecithin (disaturated PC, predominantly DPPC) and PG in lavage fluid from ARDS patients. In comparison, the relative concentrations of SM, PS, and PI fractions were much higher. Moreover, low levels of lecithin/sphingomyelin ratio (<2) and PG (<1% of total phospholipids) were consistently associated with respiratory failure [25].
Following these findings, a study evaluated lavage fluid phospholipid composition in trauma-related ARDS and classified patients according to the severity of respiratory failure [26]. This study demonstrated a correlation progressive decrease in PC composition and severity of respiratory failure, suggesting that the lower fractional PC composition is related to the severity of ARDS [26]. When trauma patients with respiratory failure developed sepsis, there were significant perturbations in phospholipid distribution in the alveolar fluid, with a substantial increase in PE combined with lower levels of lavage PC [27]. The surfactant biophysical perturbations of altered surface activity, low PC and PG with increased PI, SM, PS and PE are also seen in patients at-risk of developing ARDS, suggesting that early surfactant supplementation may mitigate ARDS progression [28].
The total lavage phospholipid (PL) content is variable between studies. There are many reasons for this variability, including various degrees of inflammatory cell membrane infiltration, and variations in the sample recovery and analytical methods used [25][28][29]. The lavage PL content may also depend on when the phospholipid analysis was performed during the various stages of ARDS. When ARDS is classified as early (<36 h after clinical features and diagnosis of ARDS), intermediate (>36 h, <6 days), or late (>6 days), the total PL content is increased in the early stages, but with a marked reduction in concentrations at the later stages of ARDS [30]. It is important to recognize that recovered lung fluid from ARDS patients contains membrane material other than lung surfactant, predominately extracellular vesicles from the increased airway neutrophil concentration. Consequently, the increased fractional concentrations of SM and PS, characteristic of cell membranes, are most likely derived from extracellular vesicles rather than from altered surfactant composition.
A more detailed analysis of the fatty acid profile of lavage fluid showed a marked reduction in palmitic acid (16:0) and saturated fatty acids in patients with ARDS [31]. Consistent with this finding, molecular species analysis of lavage phospholipid from ARDS patients demonstrated a significant reduction in dipalmitoyl PC and increased fractional concentrations of unsaturated and polyunsaturated PC species such as PC16:0/18:2, PC16:0/18:1 and PC16:0/20:4, characteristic of cell membrane material [32][33]. The degree of oxygenation impairment correlated with DPPC levels in large aggregate fractions isolated by high-speed centrifugation and more importantly, continued phospholipid alterations were associated with adverse outcomes [32]. Consistent with this finding Nakos et al. demonstrated that lack of recovery of lavaged PC during the disease course of ARDS is associated with increased mortality [30]. This suggests that during surfactant replacement a longer duration of therapy may be required in some patients with continued surfactant deficiency.
More recently, similar to the findings of the ARDS population, significant alterations in surfactant phospholipid molecular composition with reduced PC, DPPC and PG levels with reciprocal increments in other phospholipids, were seen in COVID-19 patients with severe pneumonia and ARDS [34][35]. In summary, alveolar lavage fluid from ARDS patients demonstrates significant alterations in the functional ability to maintain surface tension, phospholipid content, distribution of phospholipid categories, and PC molecular distribution, and continued surfactant alterations are associated with adverse clinical outcomes.
2.2. Surfactant Protein Alterations in ARDS
Alterations in surfactant composition are not limited to the phospholipid fraction. Although the findings are variable between studies, in general, there are reductions in concentrations of lavage surfactant proteins SP-A, SP-B and SP-C with reciprocal increases in plasma SP-A and SP-D levels [28][32][36]. While serum SP-A and SP-B levels may predict the development of ARDS [37][38], higher plasma SP-D levels correlate with disease severity and poor outcomes [39]. These high concentrations in plasma reflect the increased alveolar permeability and the consequent leakage of SP-D into the systemic circulation.
2.3. Surfactant Extraction and Analytical Methods
Alveolar surfactant isolation, purification and quantification require invasive procedures such as bronchoalveolar lavage (BAL), which, although a safe procedure, requires medical personnel to perform the procedure. Patients often require additional sedation, and in mechanically ventilated patients with hypoxemic respiratory failure desaturations and change in respiratory mechanics during and after the procedure are common [40][41]. Moreover, there are significant variations in quantitative measurements due to variations in sample recovery and will depend on the total segments lavaged. A theoretical risk of further surfactant depletion and atelectasis following a large volume lavage also exists. Nevertheless, BAL has been extensively used in ARDS patients to access surfactant material. Small volume BAL is an alternative to characterize surfactant composition without further compromising the patient’s clinical condition, but limits the ability to perform quantitative measurements [25][33]. Similarly, tracheal aspirates can also minimize procedure-related complications. Studies of healthy humans suggest comparable phospholipid composition from tracheal aspirates and can be used for surfactant molecular analysis [5]. However, the limitations include variability in recovery and inability to provide quantitative measures and, in ARDS patients, the phospholipid composition may be contaminated by inflammatory cell membrane phospholipid constituents [33]. Recent advances in microparticle extraction from lungs utilizing Particles of Exhaled Air (PExA) is an alternative and attractive way to extract alveolar surfactant material non-invasively [42].
Surfactant analysis requires centrifugation to extract the surfactant pellet, followed by lipid extraction and analysis by various gas–liquid chromatography, high performance liquid chromatography (HPLC) and mass spectrometry (ESI-MS) techniques to quantify surfactant phospholipids. De novo surfactant synthesis and metabolism in humans can be characterized by isotope labelling of surfactant phospholipid components. A combination of isotope labelling with tracer kinetics modelling and mass spectrometry analytical methods is used to measure surfactant synthesis and metabolism in ARDS patients in vivo [33][43]. The tracer substances vary between studies, but essentially include deuterated choline, deuterated water, 13C-glucose, 13C-palmitate, and 13C-acetate, which all incorporate into surfactant phospholipids enabling assessment of synthesis and metabolism of endogenous surfactant de novo [44][45]. Recent advances in spectroscopic techniques can minimize the analytical time required to measure specific surfactant phospholipid components such as DPPC, bypassing the need for detailed mass spectrometry analytical steps [35][46].
3. Molecular Mechanisms of Surfactant Alterations in ARDS
The molecular mechanisms of surfactant alterations in ARDS are complex. ARDS is characterized by significant inflammatory cell infiltration and alveolar epithelial and endothelial injury. Lung infection and aspiration of gastric contents can directly damage AT-II cells and impair surfactant synthesis, secretion, and recycling. Studies of various animal models of lung injury and AT-II cells suggest variations in surfactant synthesis [47][48][49][50][51]. The conflicting results are due to the variability in the lung injury models, the dose and duration of the insult exposure, and the timing of surfactant measurements taken. Human adult studies of isotope labelling of surfactant precursors suggest that despite very low surfactant PC, DPPC, or SatPC pool sizes, there may be increased synthesis and secretion by existing AT-II cells [33][43]. This implies that surfactant synthesis may be preserved, or even increased in functional AT-II cells and other factors may contribute to the surfactant alterations seen in ARDS. Both direct and indirect injuries can increase alveolar and systemic inflammatory response leading to cellular damage. Alveolar endothelial and epithelial injury can cause an influx of protein-rich pulmonary oedema, containing inflammatory exudate, cellular debris, and plasma proteins, which can destabilize surfactant film and directly impair surfactant activity [52][53][54][55]. Increased oxidative stress from overt inflammation and alveolar hyperoxia from oxygen therapy can result in the oxidation of surfactant phospholipids and proteins [56][57]. Moreover, activation of sPLA2-mediated hydrolysis leads to surfactant phospholipid catabolism and generation of lysophosphatidylcholines, compromising the surfactant function even further [58][59][60]. While all these mechanisms can lead to alterations in surfactant composition and function, assessing relative contribution is far more complex, particularly in human in vivo clinical settings.
This entry is adapted from the peer-reviewed paper 10.3390/diagnostics13182964
References
- Batenburg, J.J. Surfactant phospholipids: Synthesis and storage. Am. J. Physiol. 1992, 262 Pt 1, L367–L385.
- Han, S.; Mallampalli, R.K. The Role of Surfactant in Lung Disease and Host Defense against Pulmonary Infections. Ann. Am. Thorac. Soc. 2015, 12, 765–774.
- Chroneos, Z.C.; Sever-Chroneos, Z.; Shepherd, V.L. Pulmonary surfactant: An immunological perspective. Cell Physiol. Biochem. 2010, 25, 13–26.
- Postle, A.D.; Heeley, E.L.; Wilton, D.C. A comparison of the molecular species compositions of mammalian lung surfactant phospholipids. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2001, 129, 65–73.
- Dushianthan, A.; Goss, V.; Cusack, R.; Grocott, M.P.; Postle, A.D. Phospholipid composition and kinetics in different endobronchial fractions from healthy volunteers. BMC Pulm. Med. 2014, 14, 10.
- Bernhard, W.; Pynn, C.J.; Jaworski, A.; Rau, G.A.; Hohlfeld, J.M.; Freihorst, J.; Poets, C.F.; Stoll, D.; Postle, A.D. Mass spectrometric analysis of surfactant metabolism in human volunteers using deuteriated choline. Am. J. Respir. Crit. Care Med. 2004, 170, 54–58.
- Possmayer, F. Biophysical activities of pulmonary surfactant. Fetal Neonatal Physiol. 1991, 2, 949–956.
- Wright, J.R. Immunoregulatory functions of surfactant proteins. Nat. Rev. Immunol. 2005, 5, 58–68.
- Whitsett, J.A.; Wert, S.E.; Weaver, T.E. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu. Rev. Med. 2010, 61, 105–119.
- Watson, A.; Madsen, J.; Clark, H.W. SP-A and SP-D: Dual Functioning Immune Molecules with Antiviral and Immunomodulatory Properties. Front. Immunol. 2021, 11, 622598.
- Sweet, D.G.; Carnielli, V.; Greisen, G.; Hallman, M.; Ozek, E.; Te Pas, A.; Plavka, R.; Roehr, C.C.; Saugstad, O.D.; Simeoni, U.; et al. European Consensus Guidelines on the Management of Respiratory Distress Syndrome—2019 Update. Neonatology 2019, 115, 432–450.
- Goss, V.; Hunt, A.N.; Postle, A.D. Regulation of lung surfactant phospholipid synthesis and metabolism. Biochim. Biophys. Acta 2013, 1831, 448–458.
- Agassandian, M.; Mallampalli, R.K. Surfactant phospholipid metabolism. Biochim. Biophys. Acta 2013, 1831, 612–625.
- Dushianthan, A.; Grocott, M.P.; Postle, A.D.; Cusack, R. Acute respiratory distress syndrome and acute lung injury. Postgrad. Med. J. 2011, 87, 612–622.
- Matthay, M.A.; Arabi, Y.M.; Siegel, E.R.; Ware, L.B.; Bos, L.D.J.; Sinha, P.; Beitler, J.R.; Wick, K.D.; Curley, M.A.Q.; Constantin, J.M.; et al. Phenotypes and personalized medicine in the acute respiratory distress syndrome. Intensive Care Med. 2020, 46, 2136–2152.
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, Patterns of Care, and Mortality for Patients with Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016, 315, 788–800.
- Bos, L.D.J.; Ware, L.B. Acute respiratory distress syndrome: Causes, pathophysiology, and phenotypes. Lancet 2022, 400, 1145–1156.
- Wang, Y.; Zhang, L.; Xi, X.; Zhou, J.X.; The China Critical Care Sepsis Trial (CCCST) Workgroup. The Association Between Etiologies and Mortality in Acute Respiratory Distress Syndrome: A Multicenter Observational Cohort Study. Front. Med. 2021, 8, 739596.
- Engelhardt, L.J.; Olbricht, C.; Niemann, M.; Graw, J.A.; Hunsicker, O.; Weiss, B.; Bunger, V.; Weber-Carstens, S.; Boie, S.D.; Piper, S.K.; et al. Outcome Comparison of Acute Respiratory Distress Syndrome (ARDS) in Patients with Trauma-Associated and Non-Trauma-Associated ARDS: A Retrospective 11-Year Period Analysis. J. Clin. Med. 2022, 11, 5734.
- Yang, P.; Formanek, P.; Scaglione, S.; Afshar, M. Risk factors and outcomes of acute respiratory distress syndrome in critically ill patients with cirrhosis. Hepatol. Res. 2019, 49, 335–343.
- Doyle, H.R.; Marino, I.R.; Miro, A.; Scott, V.; Martin, M.; Fung, J.; Kramer, D.; Starzl, T.E. Adult respiratory distress syndrome secondary to end-stage liver disease-successful outcome following liver transplantation. Transplantation 1993, 55, 292–296.
- Wilson, J.G.; Calfee, C.S. ARDS Subphenotypes: Understanding a Heterogeneous Syndrome. Crit. Care 2020, 24, 102.
- Martin, T.R.; Zemans, R.L.; Ware, L.B.; Schmidt, E.P.; Riches, D.W.H.; Bastarache, L.; Calfee, C.S.; Desai, T.J.; Herold, S.; Hough, C.L.; et al. New Insights into Clinical and Mechanistic Heterogeneity of the Acute Respiratory Distress Syndrome: Summary of the Aspen Lung Conference 2021. Am. J. Respir. Cell Mol. Biol. 2022, 67, 284–308.
- Petty, T.L.; Silvers, G.W.; Paul, G.W.; Stanford, R.E. Abnormalities in lung elastic properties and surfactant function in adult respiratory distress syndrome. Chest 1979, 75, 571–574.
- Hallman, M.; Spragg, R.; Harrell, J.H.; Moser, K.M.; Gluck, L. Evidence of lung surfactant abnormality in respiratory failure. Study of bronchoalveolar lavage phospholipids, surface activity, phospholipase activity, and plasma myoinositol. J. Clin. Investig. 1982, 70, 673–683.
- Pison, U.; Seeger, W.; Buchhorn, R.; Joka, T.; Brand, M.; Obertacke, U.; Neuhof, H.; Schmit-Neuerburg, K.P. Surfactant abnormalities in patients with respiratory failure after multiple trauma. Am. Rev. Respir. Dis. 1989, 140, 1033–1039.
- Pison, U.; Obertacke, U.; Brand, M.; Seeger, W.; Joka, T.; Bruch, J.; Schmit-Neuerburg, K.P. Altered pulmonary surfactant in uncomplicated and septicemia-complicated courses of acute respiratory failure. J. Trauma 1990, 30, 19–26.
- Gregory, T.J.; Longmore, W.J.; Moxley, M.A.; Whitsett, J.A.; Reed, C.R.; Fowler, A.A., 3rd; Hudson, L.D.; Maunder, R.J.; Crim, C.; Hyers, T.M. Surfactant chemical composition and biophysical activity in acute respiratory distress syndrome. J. Clin. Investig. 1991, 88, 1976–1981.
- Gunther, A.; Siebert, C.; Schmidt, R.; Ziegler, S.; Grimminger, F.; Yabut, M.; Temmesfeld, B.; Walmrath, D.; Morr, H.; Seeger, W. Surfactant alterations in severe pneumonia, acute respiratory distress syndrome, and cardiogenic lung edema. Am. J. Respir. Crit. Care Med. 1996, 153, 176–184.
- Nakos, G.; Kitsiouli, E.I.; Tsangaris, I.; Lekka, M.E. Bronchoalveolar lavage fluid characteristics of early intermediate and late phases of ARDS. Alterations in leukocytes, proteins, PAF and surfactant components. Intensive Care Med. 1998, 24, 296–303.
- Schmidt, R.; Meier, U.; Yabut-Perez, M.; Walmrath, D.; Grimminger, F.; Seeger, W.; Gunther, A. Alteration of fatty acid profiles in different pulmonary surfactant phospholipids in acute respiratory distress syndrome and severe pneumonia. Am. J. Respir. Crit. Care Med. 2001, 163, 95–100.
- Schmidt, R.; Markart, P.; Ruppert, C.; Wygrecka, M.; Kuchenbuch, T.; Walmrath, D.; Seeger, W.; Guenther, A. Time-dependent changes in pulmonary surfactant function and composition in acute respiratory distress syndrome due to pneumonia or aspiration. Respir. Res. 2007, 8, 55.
- Dushianthan, A.; Goss, V.; Cusack, R.; Grocott, M.P.; Postle, A.D. Altered molecular specificity of surfactant phosphatidycholine synthesis in patients with acute respiratory distress syndrome. Respir. Res. 2014, 15, 128.
- Postle, A.D.; Clark, H.W.; Fink, J.; Madsen, J.; Koster, G.; Panchal, M.; Djukanovic, R.; Brealey, D.; Grocott, M.P.W.; Dushianthan, A. Rapid Phospholipid Turnover after Surfactant Nebulization in Severe COVID-19 Infection: A Randomized Clinical Trial. Am. J. Respir. Crit. Care Med. 2022, 205, 471–473.
- Schousboe, P.; Ronit, A.; Nielsen, H.B.; Benfield, T.; Wiese, L.; Scoutaris, N.; Verder, H.; Berg, R.M.G.; Verder, P.; Plovsing, R.R. Reduced levels of pulmonary surfactant in COVID-19 ARDS. Sci. Rep. 2022, 12, 4040.
- Greene, K.E.; Wright, J.R.; Steinberg, K.P.; Ruzinski, J.T.; Caldwell, E.; Wong, W.B.; Hull, W.; Whitsett, J.A.; Akino, T.; Kuroki, Y.; et al. Serial changes in surfactant-associated proteins in lung and serum before and after onset of ARDS. Am. J. Respir. Crit. Care Med. 1999, 160, 1843–1850.
- Greene, K.E.; Ye, S.; Mason, R.J.; Parsons, P.E. Serum surfactant protein-A levels predict development of ARDS in at-risk patients. Chest 1999, 116 (Suppl. S1), 90S–91S.
- Bersten, A.D.; Hunt, T.; Nicholas, T.E.; Doyle, I.R. Elevated plasma surfactant protein-B predicts development of acute respiratory distress syndrome in patients with acute respiratory failure. Am. J. Respir. Crit. Care Med. 2001, 164, 648–652.
- Eisner, M.D.; Parsons, P.; Matthay, M.A.; Ware, L.; Greene, K.; Acute Respiratory Distress Syndrome Network. Plasma surfactant protein levels and clinical outcomes in patients with acute lung injury. Thorax 2003, 58, 983–988.
- Steinberg, K.P.; Mitchell, D.R.; Maunder, R.J.; Milberg, J.A.; Whitcomb, M.E.; Hudson, L.D. Safety of bronchoalveolar lavage in patients with adult respiratory distress syndrome. Am. Rev. Respir. Dis. 1993, 148, 556–561.
- Klein, U.; Karzai, W.; Zimmermann, P.; Hannemann, U.; Koschel, U.; Brunner, J.X.; Remde, H. Changes in pulmonary mechanics after fiberoptic bronchoalveolar lavage in mechanically ventilated patients. Intensive Care Med. 1998, 24, 1289–1293.
- Hussain-Alkhateeb, L.; Bake, B.; Holm, M.; Emilsson, O.; Mirgorodskaya, E.; Olin, A.C. Novel non-invasive particles in exhaled air method to explore the lining fluid of small airways-a European population-based cohort study. BMJ Open Respir. Res. 2021, 8, e000804.
- Simonato, M.; Baritussio, A.; Ori, C.; Vedovelli, L.; Rossi, S.; Dalla Massara, L.; Rizzi, S.; Carnielli, V.P.; Cogo, P.E. Disaturated-phosphatidylcholine and surfactant protein-B turnover in human acute lung injury and in control patients. Respir. Res. 2011, 12, 36.
- Carnielli, V.P.; Zimmermann, L.J.; Hamvas, A.; Cogo, P.E. Pulmonary surfactant kinetics of the newborn infant: Novel insights from studies with stable isotopes. J. Perinatol. 2009, 29 (Suppl. S2), S29–S37.
- Brandsma, J.; Bailey, A.P.; Koster, G.; Gould, A.P.; Postle, A.D. Stable isotope analysis of dynamic lipidomics. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 792–796.
- Ahmed, W.; Veluthandath, A.V.; Rowe, D.J.; Madsen, J.; Clark, H.W.; Postle, A.D.; Wilkinson, J.S.; Murugan, G.S. Prediction of Neonatal Respiratory Distress Biomarker Concentration by Application of Machine Learning to Mid-Infrared Spectra. Sensors 2022, 22, 1744.
- Lewis, J.F.; Ikegami, M.; Jobe, A.H. Altered surfactant function and metabolism in rabbits with acute lung injury. J. Appl. Physiol. 1990, 69, 2303–2310.
- Gross, N.J. Surfactant subtypes in experimental lung damage: Radiation pneumonitis. Am. J. Physiol. 1991, 260 Pt 1, L302–L310.
- Holm, B.A.; Matalon, S.; Finkelstein, J.N.; Notter, R.H. Type II pneumocyte changes during hyperoxic lung injury and recovery. J. Appl. Physiol. 1988, 65, 2672–2678.
- Crim, C.; Longmore, W.J. Sublethal hydrogen peroxide inhibits alveolar type II cell surfactant phospholipid biosynthetic enzymes. Am. J. Physiol. 1995, 268 Pt 1, L129–L135.
- Kennedy, K.A.; Snyder, J.M.; Stenzel, W.; Saito, K.; Warshaw, J.B. Vitamin E alters alveolar type II cell phospholipid synthesis in oxygen and air. Exp. Lung Res. 1990, 16, 607–615.
- Holm, B.A.; Wang, Z.; Notter, R.H. Multiple mechanisms of lung surfactant inhibition. Pediatr. Res. 1999, 46, 85–93.
- Moses, D.; Holm, B.A.; Spitale, P.; Liu, M.Y.; Enhorning, G. Inhibition of pulmonary surfactant function by meconium. Am. J. Obstet. Gynecol. 1991, 164, 477–481.
- Seeger, W.; Grube, C.; Gunther, A.; Schmidt, R. Surfactant inhibition by plasma proteins: Differential sensitivity of various surfactant preparations. Eur. Respir. J. 1993, 6, 971–977.
- Echaide, M.; Autilio, C.; Arroyo, R.; Perez-Gil, J. Restoring pulmonary surfactant membranes and films at the respiratory surface. Biochim. Biophys. Acta Biomembr. 2017, 1859 Pt B, 1725–1739.
- Putman, E.; van Golde, L.M.; Haagsman, H.P. Toxic oxidant species and their impact on the pulmonary surfactant system. Lung 1997, 175, 75–103.
- Rodriguez-Capote, K.; Manzanares, D.; Haines, T.; Possmayer, F. Reactive oxygen species inactivation of surfactant involves structural and functional alterations to surfactant proteins SP-B and SP-C. Biophys. J. 2006, 90, 2808–2821.
- Arbibe, L.; Koumanov, K.; Vial, D.; Rougeot, C.; Faure, G.; Havet, N.; Longacre, S.; Vargaftig, B.B.; Bereziat, G.; Voelker, D.R.; et al. Generation of lyso-phospholipids from surfactant in acute lung injury is mediated by type-II phospholipase A2 and inhibited by a direct surfactant protein A-phospholipase A2 protein interaction. J. Clin. Investig. 1998, 102, 1152–1160.
- Chabot, S.; Koumanov, K.; Lambeau, G.; Gelb, M.H.; Balloy, V.; Chignard, M.; Whitsett, J.A.; Touqui, L. Inhibitory effects of surfactant protein A on surfactant phospholipid hydrolysis by secreted phospholipases A2. J. Immunol. 2003, 171, 995–1000.
- Holm, B.A.; Keicher, L.; Liu, M.Y.; Sokolowski, J.; Enhorning, G. Inhibition of pulmonary surfactant function by phospholipases. J. Appl. Physiol. 1991, 71, 317–321.
This entry is offline, you can click here to edit this entry!