While it is true that pharmacotherapy has achieved desired health outcomes, significant unmet medical needs persist in the field of central nervous system (CNS) drugs, particularly for neurodegenerative diseases such as Alzheimer’s disease, as well as ocular diseases such as diabetic retinopathy and age-related macular degeneration. Drugs cannot enter the brain from the bloodstream due to the presence of the blood–brain barrier (BBB). Similarly, they cannot enter the eyes from the bloodstream due to the blood–retina barrier (BRB), which is composed of the endothelium or the epithelium. Thus, innovative drug delivery systems that can overcome these barriers based on efflux transporters, hydrophobic lipid bilayer membranes, and tight junctions should be developed using patient-friendly techniques distinct from craniotomy procedures or intravitreal injections. Brain-penetrating CNS drugs and antihistamine drugs commonly share N-containing groups. These findings suggest that certain types of cation transporters are involved in their transportation across the cell membrane.
- blood–brain barrier
- the blood–retina barrier
- drug delivery system
- transmembrane drug delivery
1. Introduction
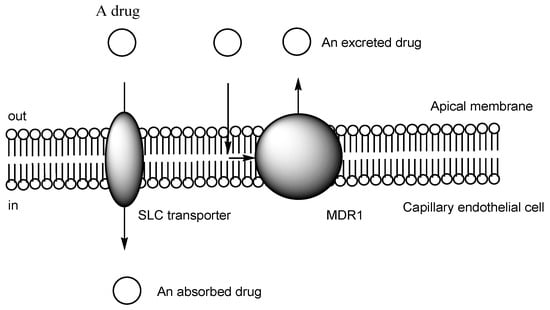
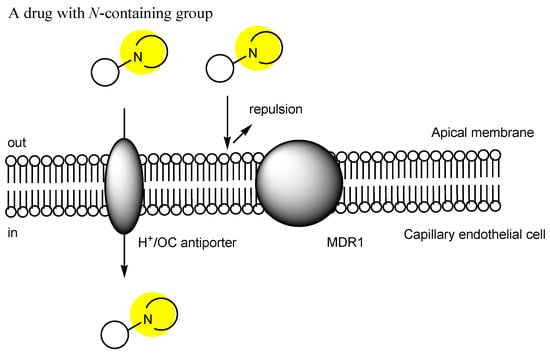
2. Transporter-Conscious Drug Design
| # | Categories | Transporters/Subtypes | Substrates |
|---|---|---|---|
| (1) | Amine transporters | The proton-coupled organic cation (H+/OC) antiporter, organic cation transporter novel type 1 (OCTN1), OCTN2, OCTN3, multidrug and toxin extrusion protein 1 (MATE1), MATE2, MATE3, plasma membrane monoamine transporter (PMAT) | Cationic amine compounds |
| (2) | Peptide transporters | Peptide transporter 1 (PEPT1), PEPT2 | Peptides |
| (3) | Amino acid transporters | L-type amino acid transporter 1 (LAT1), LAT2, LAT3, LAT4 | Amino acids |
| (4) | Organic cation transporters (OCTs) | OCT1, OCT2, OCT3, OCT4 | Cationic compounds |
| (5) | Organic anion transporters (OATs) | OAT1, OAT2, OAT3, OAT4, OAT5, organic anion transporting peptides (OATP1A2), OATP1B1, OATP1B3, OATP1C1 OATP2A1, OATP2B1, OATP3A1, OATP4A1, OATP4C1, OATP5A1, OATP6A1 |
Anionic compounds |
| (6) | Glucose transporters | Glucose transporter1 (GLUT1), GLUT2, GLUT3, GLUT4, GLUT5, GLUT6, GLUT7 | Glucose |
3. Implementation of Transporter-Conscious Drug Design with N-Containing Groups
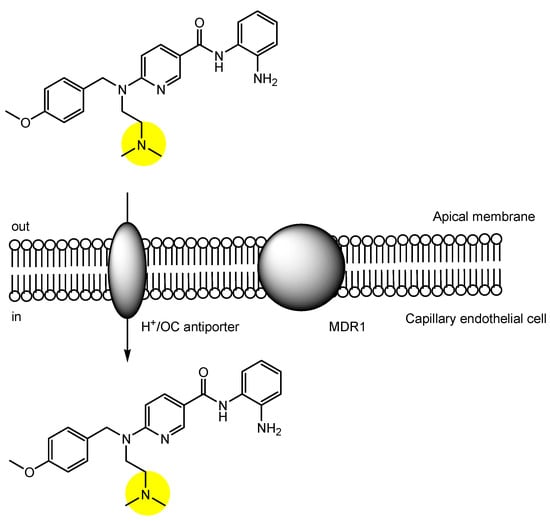
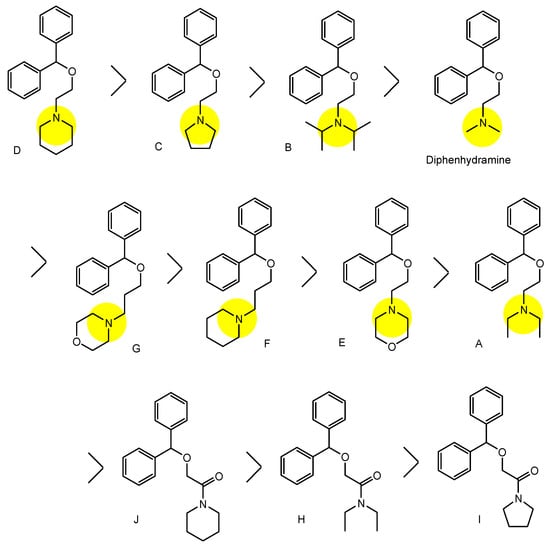
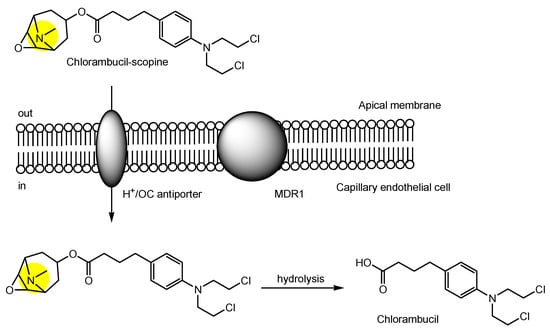
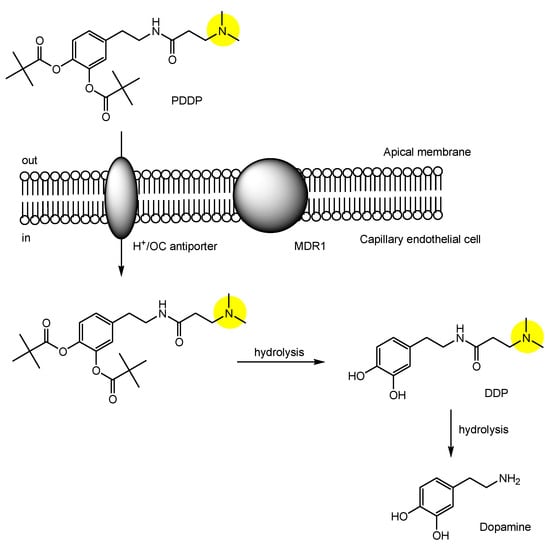
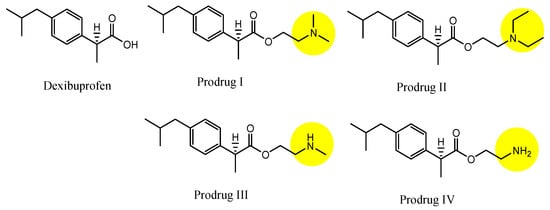
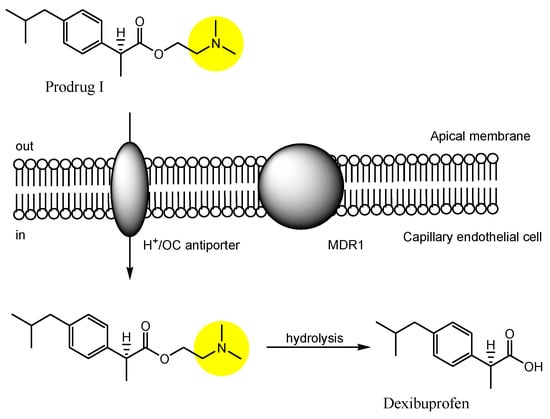
This entry is adapted from the peer-reviewed paper 10.3390/futurepharmacol3040046
References
- Stimulus package. Nat. Med. 2018, 24, 247.
- Angermann, R.; Rauchegger, T.; Nowosielski, Y.; Casazza, M.; Bilgeri, A.; Ulmer, H.; Zehetner, C. Treatment compliance and adherence among patients with diabetic retinopathy and age-related macular degeneration treated by anti-vascular endothelial growth factor under universal health coverage. Graefe’s Arch. Clin. Exp. Ophthalmol. 2019, 257, 2119–2125.
- Tashima, T. Smart Strategies for Therapeutic Agent Delivery into Brain across the Blood-Brain Barrier Using Receptor-Mediated Transcytosis. Chem. Pharm. Bull. 2020, 68, 316–325.
- Tashima, T. Intriguing possibilities and beneficial aspects of transporter-conscious drug design. Bioorg. Med. Chem. 2015, 23, 4119–4131.
- Tashima, T. Intelligent substance delivery into cells using cell-penetrating peptides. Bioorg. Med. Chem. Lett. 2017, 27, 121–130.
- Tashima, T. Effective cancer therapy based on selective drug delivery into cells across their membrane using receptor-mediated endocytosis. Bioorg. Med. Chem. Lett. 2018, 28, 3015–3024.
- Tashima, T. Shortcut Approaches to Substance Delivery into the Brain Based on Intranasal Administration Using Nanodelivery Strategies for Insulin. Molecules 2020, 25, 5188.
- Tashima, T. Delivery of Intravenously Administered Antibodies Targeting Alzheimer’s Disease-Relevant Tau Species into the Brain Based on Receptor-Mediated Transcytosis. Pharmaceutics 2022, 14, 411.
- Tashima, T. Brain Cancer Chemotherapy through a Delivery System across the Blood-Brain Barrier into the Brain Based on Receptor-Mediated Transcytosis Using Monoclonal Antibody Conjugates. Biomedicines 2022, 10, 1597.
- Tashima, T. Delivery of Drugs into Cancer Cells Using Antibody–Drug Conjugates Based on Receptor-Mediated Endocytosis and the Enhanced Permeability and Retention Effect. Antibodies 2022, 11, 78.
- de Mora, F.; Balsa, A.; Cornide-Santos, M.; Carrascosa, J.M.; Marsal, S.; Gisbert, J.P.; Abad, M.-A.; Duarte, R.F.; Wiechmann, M.; Martínez, R. Biosimilar and interchangeable: Inseparable scientific concepts? Br. J. Clin. Pharmacol. 2019, 85, 2460–2463.
- Zamek-Gliszczynski, M.J.; Taub, M.E.; Chothe, P.P.; Chu, X.; Giacomini, K.M.; Kim, R.B.; Ray, A.S.; Stocker, S.L.; Unadkat, J.D.; Wittwer, M.B.; et al. Transporters in Drug Development: 2018 ITC Recommendations for Transporters of Emerging Clinical Importance. Clin. Pharmacol. Ther. 2018, 104, 890–899.
- Jaramillo, A.C.; Saig, F.A.; Cloos, J.; Jansen, G.; Peters, G.J. How to overcome ATP-binding cassette drug efflux transporter-mediated drug resistance? Cancer Drug Resist. 2018, 1, 6–29.
- Hu, C.; Tao, L.; Cao, X.; Chen, L. The solute carrier transporters and the brain: Physiological and pharmacological implications . Asian J. Pharm. Sci. 2020, 15, 131–144.
- Gotfryd, K.; Boesen, T.; Mortensen, J.S.; Khelashvili, G.; Quick, M.; Terry, D.S.; Missel, J.W.; LeVine, M.V.; Gourdon, P.; Blanchard, S.C.; et al. X-ray structure of LeuT in an inward-facing occluded conformation reveals mechanism of substrate release. Nat. Commun. 2020, 11, 1005.
- Kumar, S.; Athreya, A.; Gulati, A.; Nair, R.M.; Mahendran, I.; Ranjan, R.; Penmatsa, A. Structural basis of inhibition of a transporter from Staphylococcus aureus, NorC, through a single-domain camelid antibody. Commun. Biol. 2021, 4, 836.
- Roberts, A.G. The Structure and Mechanism of Drug Transporters. Methods Mol. Biol. 2021, 2342, 193–234.
- Debaisieux, S.; Rayne, F.; Yezid, H.; Beaumelle, B. The Ins and Outs of HIV-1 Tat. Traffic 2012, 13, 355–363.
- Hiranaka, S.; Tega, Y.; Higuchi, K.; Kurosawa, T.; Deguchi, Y.; Arata, M.; Ito, A.; Yoshida, M.; Nagaoka, Y.; Sumiyoshi, T. Design, Synthesis, and Blood-Brain Barrier Transport Study of Pyrilamine Derivatives as Histone Deacetylase Inhibitors. ACS Med. Chem. Lett. 2018, 9, 884–888.
- Tega, Y.; Tabata, H.; Kurosawa, T.; Kitamura, A.; Itagaki, F.; Oshitari, T.; Deguchi, Y. Structural Requirements for Uptake of Diphenhydramine Analogs into hCMEC/D3 Cells Via the Proton-Coupled Organic Cation Antiporter. J. Pharm. Sci. 2021, 110, 397–403.
- Wang, X.; Qi, B.; Su, H.; Li, J.; Sun, X.; He, Q.; Fu, Y.; Zhang, Z. Pyrilamine-sensitive proton-coupled organic cation (H+/OC) antiporter for brain-specific drug delivery. J. Control. Release 2017, 254, 34–43.
- Wang, X.; Li, J.; Xu, C.; Li, Y.; Gong, T.; Sun, X.; Fu, Y.; He, Q.; Zhang, Z. Scopine as a novel brain-targeting moiety enhances the brain uptake of chlorambucil. Bioconjug. Chem. 2014, 25, 2046–2054.
- Li, Y.; Zhou, Y.; Qi, B.; Gong, T.; Sun, X.; Fu, Y.; Zhang, Z. Brain-Specific Delivery of Dopamine Mediated by N,N-Dimethyl Amino Group for the Treatment of Parkinson’s Disease. Mol. Pharm. 2014, 11, 3174–3185.
- Li, Y.; Zhou, Y.; Jiang, J.; Wang, X.; Fu, Y.; Gong, T.; Sun, X.; Zhang, Z. Mechanism of brain targeting by dexibuprofen prodrugs modified with ethanolamine-related structures. J. Cereb. Blood Flow Metab. 2015, 35, 1985–1994.
- Huttunen, J.; Adla, S.K.; Markowicz-Piasecka, M.; Huttunen, K.M. Increased/Targeted Brain (Pro)Drug Delivery via Utilization of Solute Carriers (SLCs). Pharmaceutics 2022, 14, 1234.
- Kawase, A.; Chuma, T.; Irie, K.; Kazaoka, A.; Kakuno, A.; Matsuda, N.; Shimada, H.; Iwaki, M. Increased penetration of diphenhydramine in brain via proton-coupled organic cation antiporter in rats with lipopolysaccharide-induced inflammation. Brain Behav. Immun. Health 2021, 10, 100188.
- Kawase, A.; Kazaoka, A.; Shimada, H.; Iwaki, M. Increased brain penetration of diphenhydramine and memantine in rats with adjuvant-induced arthritis. Brain Res. 2021, 1768, 147581.
- Chapy, H.; Goracci, L.; Vayer, P.; Parmentier, Y.; Carrupt, P.A.; Declèves, X.; Scherrmann, J.M.; Cisternino, S.; Cruciani, G. Pharmacophore-based discovery of inhibitors of a novel drug/proton antiporter in human brain endothelial hCMEC/D3 cell line. Br. J. Pharmacol. 2015, 172, 4888–4904.
- Smirnova, M.; Goracci, L.; Cruciani, G.; Federici, L.; Declèves, X.; Chapy, H.; Cisternino, S. Pharmacophore-Based Discovery of Substrates of a Novel Drug/Proton-Antiporter in the Human Brain Endothelial hCMEC/D3 Cell Line. Pharmaceutics 2022, 14, 255.
- Lombardo, S.M.; Schneider, M.; Türeli, A.E.; Günday Türeli, N. Key for crossing the BBB with nanoparticles: The rational design. Beilstein J. Nanotechnol. 2020, 11, 866–883.