1. Introduction
Cell adhesion molecules (CAMs) placed on the cell surface perform critical functions in a number of biological processes requiring contact between cells or with the extracellular matrix, such as cell recognition, adhesion, migration, and differentiation. The principal CAM groups are integrins, selectins, cadherins, and the immunoglobulin superfamily. In the developing brain, CAMs are crucial for the correct assembly of synaptic connections as well as the interactions with supporting glial cells. In the mature brain, complex functions depend on the correct performance of CAMs in establishing contacts between neuronal bodies, axonal interactions with myelinating glial cells, axon fasciculation, and connections to non-nervous cells [
1,
2]. Integrins are type I transmembrane proteins consisting of a large multidomain extracellular portion, a single-pass transmembrane region, and a short cytoplasmic component. The integrin family encompasses heterodimers of α and β subunits, which can combine to form several different integrins exhibiting overlapping but non-redundant functions, with specific ligand and signalling preferences depending on the α and β subunit combinations [
3,
4]. Selectins consist of an N-terminal carbohydrate-recognition domain allowing for the binding to glycoconjugates, an epidermal growth factor-like domain, a series of short consensus repeats, a transmembrane region, and a short C-terminal intracellular tail. Three selectin family members exist, P-selectin, L-selectin, and E-selectin, expressed in platelet-endothelial cells, leukocytes, and endothelial cells, respectively [
5,
6]. In the brain, selectins expressed by endothelial cells are involved in inflammatory responses, in damage after ischemic events, and in autoimmune diseases [
6,
7,
8]. Cadherins are a large number of calcium-dependent adhesion proteins. Their structure comprises a calcium-binding extracellular domain consisting of several cadherin repeats of about 100 amino acids, a transmembrane domain, and a cytoplasmic domain which interacts with signalling molecules. Cadherins can form lateral dimers (cis-dimers) as well as trans-dimers with cadherins expressed by other cells [
9,
10].
Immunoglobulin (Ig)-like molecules are an ancient and diverse family of proteins performing a variety of functions, such as immune and signalling molecules and CAMs. Four different subtypes of Ig-like domains exist which are named constant 1, constant 2, variable, and intermediate (C1, C2, V, and I) for their resemblance to immunoglobulin domains. The presence of at least one Ig-like domain is a requirement of this class [
11,
12]. Generally, the core of all Ig domains encompasses two β-sheets facing each other and an intra-chain disulfide bridge which provides stability to the structure [
13]. In addition, variable numbers of fibronectin type III domains or other protein modules characterise the sub-families within this large group. Ig-like CAMs generally include type I transmembrane proteins with a large N-terminal domain, a transmembrane portion, and a cytoplasmic domain [
1,
14]. However, some CAMs are anchored to the membrane through a glycosylphosphatidylinositol (GPI) segment [
15]. Some proteins demonstrate homophilic binding specificity, whereas others have heterophilic specificity, thus interacting with other Ig-like CAMs or with surface proteins [
1,
16]. Ig domains play critical roles in mediating homophilic and heterophilic interactions in
trans, namely between CAMs on adjacent cells or CAMs localised to the extracellular environment, as well as in
cis, i.e., with proteins located in the plasma membrane of the same cell [
17,
18]. The first Ig-like CAMs identified in the central nervous system (CNS) were neural cell adhesion molecule (NCAM) and L1, which, along with their isoforms, are recognised for their major functions in axon outgrowth and fasciculation, neuronal survival and migration, synapse formation, and synaptic plasticity [
11,
14,
19]. Subsequent research led to the identification of additional sub-families, such as the nectins, MAM domain–containing GPI anchors, IGSF9, IGSF21, contactins, and IgLONs [
11,
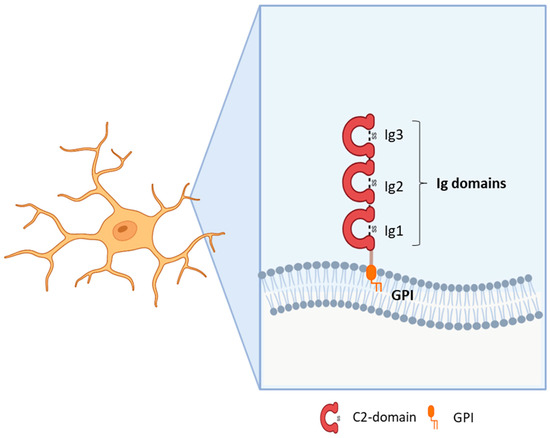
20]. The IgLON subfamily, includes five members sharing the presence of three C2 immunoglobulin domains in the N-terminus and a GPI-anchoring to the membrane (
Figure 1). The first family members identified were Limbic System Associated Membrane Protein (LSAMP, initially named LAMP) [
21], Opioid Binding Protein/Cell Adhesion Molecule Like (OPCML, also known as OBCAM) [
22], and neurotrimin (NTM) [
23]. Indeed, the name IgLON stands for Ig family containing LAMP, OBCAM, NTM. Subsequently, another family member was identified initially as KILON/neurotractin, and later re-named Neuronal Growth Regulator 1 (NEGR1) [
24,
25]. Lastly, antibodies against the IgLON member IgLON5 (IgLON Family Member 5) have been associated with autoimmune encephalitis [
26]. Since the discovery of this family, its prominent roles in brain development, axon fasciculation, neurite extension, and synapse formation and maintenance have been identified [
21,
22,
23,
24,
25]. Although the role in synapse regulation is well-established, further research is needed to characterise the molecular mechanisms underlying the functional outcomes [
17,
27,
28,
29,
30]. A meaningful contribution to the comprehension of IgLONs’ physiological functions derived from the analysis of its molecular evolution, originating from an ancestor around the emergence of Arthropods [
31]. Functional analysis allowed for the identification of common as well as specific sequences for interacting partner recognition, post-translational modifications, metalloproteinase cleavage sites, and signal transduction pathways [
31].
Figure 1. Structure of IgLON family members.
Available evidence supports the involvement of CAMs in the onset of neuropsychiatric disorders. In Alzheimer’s disease, a role for CAMs has been suggested by genome-wide association studies (GWAS) as well as by the identification of altered levels in diseased brains [
32,
33]. Molecules belonging to several CAM families have been implicated in pathological manifestations through different mechanisms, involving amyloid-β metabolism, cell plasticity, and neuroinflammation [
33,
34]. The detection of an increase in the enzymes involved in GPI-anchoring in Alzheimer’s disease brains provided further support to a relevant role of these sub-families [
35]. Moreover, genetic studies have associated CAMs with a number of neuropsychiatric disorders, including CRASH syndrome, MASA syndrome, X-linked mental retardation, intellectual disability, autism spectrum disorder, schizophrenia, addiction, and bipolar disorder [
15,
19]. The roles of CAMs in axon growth, guidance, and fasciculation; in target recognition; and in synapse formation and maintenance might contribute to the circuit alterations characterising neuropsychiatric disorders. The main proposed mechanisms include the ability to target the formation of specific circuitries responsible for specialised brain functions through localised expression. Additionally, CAMs may impact the occurrence of neuropsychiatric diseases by modifying the balance between excitatory and inhibitory signals, thereby affecting local neural circuit activity. This influence extends to the modulation of neuromodulatory systems like monoaminergic circuits and the alteration of synaptic strength and composition [
15]. Focussing more specifically on Ig-like CAMs, mutations in the genes belonging to the best-studied NCAM and L1 families, as well as changes in their expression patterns or post-translational modifications, have been associated with psychiatric and neurodegenerative disorders, including the L1 syndrome, Alzheimer’s disease, schizophrenia, and bipolar disorder [
36]. When further restricting the focus to GPI-anchored sub-families, their role as functional receptors controlling neurite outgrowth, synapse formation, synapse plasticity, and learning behaviours has been implicated in the pathophysiological mechanisms associated with neurodegenerative and psychiatric disorders [
17].
2. IgLON5 (IgLON Family Member 5)
Anti-IgLON5 disease (ORPHA:420789) is a rare disease of the CNS. Since its discovery in 2014, 60 cases have been reported worldwide [
37,
38,
39]. The clinical manifestation of the disease is highly heterogeneous, encompassing unique sleep and movement disorders, bulbar dysfunction, and cognitive impairment [
26]. Disease onset is around the age of 60 years with progressive symptoms that can culminate in life-threatening respiratory problems [
37]. Post-mortem analysis has revealed a tauopathy restricted to neurons in the brainstem, tegmentum, hypothalamus, and hippocampus [
40]. The mortality rate associated with the anti-IgLON5 syndrome is considerably high, contributing to an overall mortality rate of 34%, with no discernible link between mortality and treatment response [
38]. While respiratory complications are the primary cause of death, the additional neurological symptoms (cognitive decline, sleep disturbances) significantly diminish the quality of life for patients and their families.
The role of the IgLON5 protein and the underlying mechanism of disease associated with anti-IgLON5 antibodies remain poorly understood. IgLON5 is widely expressed in the CNS, with the highest levels reached in the olfactory bulb, cortical plate, and hippocampus in mice and humans. Comprising a chain of 336 amino acids, IgLON5 shares significant structural resemblance with other proteins such as OPCML (50% similarity), NTM (48–49% similarity), LSAMP (46–47% similarity), and NEGR1 (41% similarity) (
Figure 1). IgLON5 is involved in forming both homomeric and heteromeric interactions with other IgLON family members [
41] (
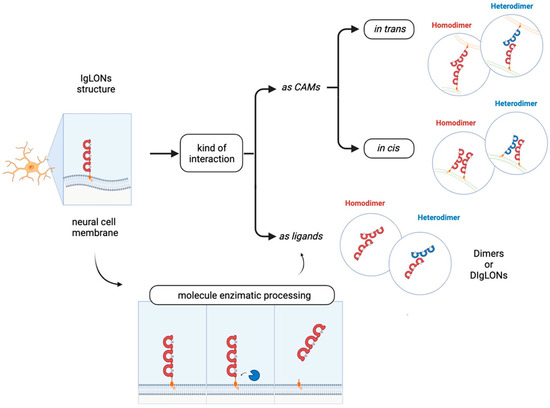
Figure 2). However, the functional implications stemming from these interactions remain poorly characterised.
Figure 2. IgLONs can form homo- and hetero-dimers to interact in trans (in different cells) or in cis (in the same cell). They can be cleaved from the membrane by metalloproteases to form soluble interacting proteins.
The detection of anti-IgLON5 antibodies is crucial for the diagnosing of anti-IgLON5 disease. These antibodies are detectable in both serum and cerebrospinal fluid (CSF), typically coinciding with the initial diagnosis timeframe. The question of whether anti-IgLON5 antibodies directly cause neuronal dysfunction and degeneration or instead appear as a consequence of the neurodegenerative process remain unresolved. Among anti-IgLON5 antibodies, the non-complement fixing IgG4 subclass predominates over IgG1. In vitro experiments have shown that IgG4 antibodies lead to the internalization of IgLON5 in cultures of hippocampal neurons [
42]. Upon exposure to anti-IgLON5 antibodies, cultured rat or human neurons showed increased neurodegenerative features such as neuronal blebbing and fragmentation [
43,
44]. Prolonged exposure to anti-IgLON5 IgG prompts tau hyperphosphorylation and cellular death [
44].
Similar to Alzheimer’s disease, the tau filaments evident in individuals with anti-IgLON5 syndrome encompass both 3R-tau and 4R-tau isoforms. However, this accumulation manifests in a distinct spatial pattern, indicative of anti-IgLON5 disease constituting a novel form of tauopathy. Recent autoptic studies have described anti-IgLON5 cases lacking hyperphosphorylated tau deposits [
45,
46]. These findings raise the possibility that tauopathy might arise subsequently in the disease progression, evolving as a prolonged outcome of the antibody-related effects. Thus, earlier events still depending on anti-IgLON5 antibodies may play a role in the aetiology of the disease, in addition to the proposition that a genetic predisposition to autoimmunity might exert influence, exemplified by the robust correlation with the exceptionally rare HLA-DRB1*1001 and HLA-DQB1*0501 alleles [
40].
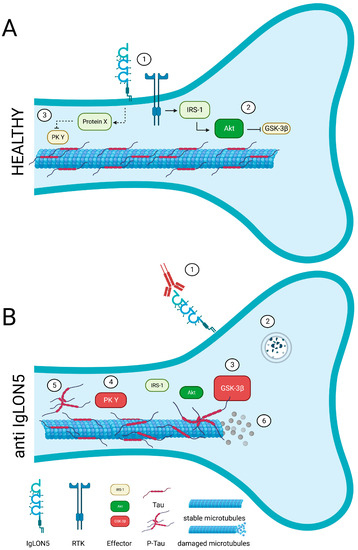
IgLONs interact and regulate receptor tyrosine kinase (RTK) trafficking and, eventually, their signalling impinging on ERK1/2 and AKT phosphorylation [
28,
29]. Anti-IgLON5 disease is characterised by the aggregation of hyperphosphorylated-Tau, which is phosphorylated by a variety of serine/threonine protein kinases [
47]; GSK-3𝛽 is the main kinase responsible for Tau phosphorylation and precipitation [
48]. An intricate signalling cascade, encompassing RTKs, insulin receptor substrate 1 (IRS-1), and AKT, has been identified as a key mechanism in curbing Tau phosphorylation by GSK-3𝛽 [
49]. Hence, the hypothesis that IgLON5 may interact with and regulate a yet to be identified RTK is exceptionally intriguing (
Figure 3). Recent in vitro findings put forth the proposition that antibodies against anti-IgLON5 could potentially interfere with the IgLON5 interactome [
50]. It is plausible that these anti-IgLON5 antibodies might impede the IgLON5-RTK interaction, thereby exerting an influence on RTK signalling. This sequence of events could consequently trigger aberrant Tau phosphorylation and accumulation, ultimately culminating in neuronal dysfunction (
Figure 3).
Figure 3. A potential link between IgLON5 and the aggregation of hyperphosphorylated-Tau. (A) In healthy conditions, IgLON5 may interact with and regulate a yet to be identified receptor tyrosine kinase (RTK, 1). A signalling cascade involving such RTK, insulin receptor substrate 1 (IRS-1), and AKT (2) prevents Tau phosphorylation by GSK-3β (3). A different RTK-triggered pathway may control Tau phosphorylation (4) and assure microtubule stability. (B) In patients, the anti-IgLON5 antibodies sequester IgLON5 (1) and trigger the degradation of IgLON5 together with the RTK, potentially (2). As such, RTK signalling pathways are altered (3–4). This may result in pathological tau phosphorylation and accumulation (5) and, eventually, neurotoxic microtubule disassembly (6).
In order to pave the way for effective therapeutic interventions geared towards individuals afflicted with IgLON5 deficiency disease, a comprehensive grasp of both the normal and pathological roles of IgLON5, alongside the associated signalling cascade, becomes essential.
This entry is adapted from the peer-reviewed paper 10.3390/genes14101886