MAPK (Mitogen-Activated Protein Kinase) signaling pathways are among the best studied ubiquitous regulatory cascades that respond to a wide range of extracellular hormones, growth factors, neurotransmitters and other cellular cues to control rapid cellular responses, decision making and cell fate. MAPK cascades are comprised of three or more kinases acting in series, beginning with MAPK kinase kinases (MAP3Ks) that activate MAPK kinases (MAP2Ks or MAP/ERK kinases (MEKs)), that then with high selectivity activate MAPKs, the effector kinases of the cascades. Prior to its purification or recognition of its enzymatic activity, the MAPK ERK2 (extracellular signal-regulated kinase 2) had been observed as a 42 kDa tyrosine-phosphorylated protein in lysates from growth factor-stimulated and virally transformed cells [
1]. Subsequently, what turned out to be the same protein was found as a growth factor-activated protein kinase [
2] that phosphorylated microtubule-associated protein 2 (MAP-2 Protein Kinase) and could activate ribosomal protein S6 kinase [
3]. Purification and molecular cloning identified two closely related but distinct enzymes ERK1 (p44, MAPK3) and ERK2 (MAPK1), often the more abundant [
4,
5,
6]. Other MAPKs include Stress-Activated Protein Kinases (SAPKs)/Jun-N-terminal Kinases (JNKs), p38 family kinases and ERK5/Big MAPKs (BMK), in addition to atypical MAPK pathways ERK3/4, ERK7/8 and NLK (Nemo-like kinase) [
7,
8,
9]. These MAPKs belong to the CMGC group of highly conserved serine–threonine kinases [
10]. A hallmark of the typical MAPKs is their activation by dual phosphorylation on nearby tyrosine and threonine residues in their activation loop, in what is known as the TXY motif [
11], where the amino acid X is distinct for subsets of MAPKs. In the case of ERK1/2 and ERK5, the motif is Thr-Glu-Tyr (TEY), JNKs contain Pro as the intermediate residue (TPY), and Gly for p38s (TGY) [
9] (reviewed in more detail in [
12]).
2. ERK Pathway Components
2.1. RAS
RAS (RAt Sarcoma) refers to small GTPase (G)-proteins encoded by three genes yielding proteins with molecular weights of ~21 kDa. RAS proteins contain a GTP-binding domain and a hypervariable region (HVR) which marks the following differences between them [
13]: HRAS (Harvey sarcoma viral oncogene), NRAS (neuroblastoma oncogene) and KRAS (Kirsten sarcoma viral oncogene). KRAS has two alternatively spliced forms named KRAS4A and KRAS4B. To be active, RAS proteins need to be localized at the plasma membrane or endomembranes. Lipid post-translational modifications (PTMs), farnesylation and palmitoylation, are essential for RAS association on the membrane [
14,
15]. The membrane sub-domain and subcellular localization from which the RAS signal originates determines the downstream effectors that are activated and, consequently, the cellular outcome [
16]. As GTPases, RAS proteins cycle from the GDP-loaded OFF configurations to GTP-loaded ON conformations assisted by RAS–guanine nucleotide exchange factors (RAS-GEFs) and return to OFF states facilitated by RAS–GTPase-activating proteins (RAS-GAPs) [
17]. The RAS-GEFs SOS (Son of Sevenless)1 and SOS2 are activated by receptor tyrosine kinases coordinated by the adaptor protein Grb2 [
18]. Once activated, RAS can interact with the RAS binding domain (RBD) in RAS effectors. In addition to RAF proteins, MAP3Ks in the ERK1/2 pathway, there are over fifty other RAS effectors including RALGDS, RALBP1, RIN1, TIAM1, PLCε, REPAC, RASSF1/5, and PI3K among others [
19,
20].
The four RAS proteins show around 85% of amino acid identity differing in the hypervariable region (HVR) [
21]. In spite of their similarities, manipulation of their expression or activation results in different cellular outcomes [
22,
23]. KRAS is the only family member essential for development [
24,
25,
26]. This feature depends on its locus expression pattern in development, as the protein product of HRAS knock-in in the KRAS locus gives rise to viable mice. However, a phenotype in adult life has been observed when KRAS is replaced by HRAS [
27], although it disappears by eliminating the endogenous HRAS [
28]. These findings indicate that different RAS isoforms are apparently equivalent in physiological conditions.
Despite the high amino acid identity between RAS isoforms, their nucleotide sequences are less so [
31]. The Counter laboratory proposed codon bias as a mechanism responsible for the different expression and oncogenicity of KRAS and HRAS. Even though KRAS is the most commonly mutated isoform, it is the most poorly translated due to rare codons. This group suggested that the potent oncogenicity of HRAS lead to cell cycle arrest and senescence, while the different codon usage of KRAS overcomes this issue becoming the most frequently mutated RAS in cancer [
31]. In fact, they later demonstrated that substitution of KRAS rare codons for common codons rendered tumors less aggressive [
32]. Following studies corroborated that rare codons in KRAS affect transcription due to histone modification and transcriptional activation recruitment, mRNA translation, and even protein conformation, highlighting the importance of codon usage [
33].
2.2. RAF
RAF (Rapidly Accelerated Fibrosarcoma) is the first kinase in the pathway core, serving as the MAP3K downstream of RAS [
39]. The regulatory paradigm has it that RAF is cytosolic in resting conditions and translocates to the plasma membrane to be activated by RAS upon growth factor stimulation [
40,
41,
42] (reviewed in [
43,
44]). In mammals, the RAF family consists of ARAF (65 kDa), CRAF (65 kDa) (aka RAF1), and BRAF (84 kDa, larger due to additional BRAF-specific sequence at its N-terminus) [
45], which are ~75% identical in shared segments. Also in the family are the pseudokinases KSR1 and KSR2 discussed in
Section 4.2.
RAF consists of a RAS binding domain (RBD) and a cysteine-rich domain (CRD), both in the N-terminal regulatory region of the protein and the kinase domain in the C-terminus. RAF was formerly divided into three conserved regions named CR1, CR2, and CR3. CR1 contains the RBD and the CRD, CR2 is a serine/threonine-rich domain, and CR3 refers to the kinase domain (reviewed in [
46]). CRD facilitates RAF localization at the plasma membrane [
47,
48] and together with RBD both interact with RAS [
49,
50].
Of the three isoforms, BRAF is the most frequently mutated in cancer. BRAF mutations are present in 7.7% across all cancer types [
60]. Overall, BRAF mutation prevalence in human cancers is almost 100% for hairy cell leukemia, 50% for melanoma, 45% for papillary thyroid cancer, 10% for colon cancer, and 10% for non-small cell lung cancer cases [
61]. The most frequent mutation in BRAF is V to E substitution at residue 600 (V600E), accounting for 90% of BRAF mutations [
62]. Around 200 low-frequency cancer-associated mutations have been described in BRAF [
63]. Most of them are believed to reverse the autoinhibitory conformation in the absence of upstream signaling [
64]. BRAF mutations have been classified into three groups as follows: class I mutations are those that affect BRAF V600 position, most commonly V600E but also V600K/D/R/M which are able to transduce the signal downstream as monomers; class II mutations function as RAS-independent dimers with reported increased kinase activity but still weaker than BRAF V600 mutants; and class III mutants show impaired kinase activity and do not directly phosphorylate MEK but retain the ability to bind RAS and heterodimerize with CRAF [
65,
66].
2.3. MEK
Downstream of RAF are the two ERK-specific MAP2Ks [
68,
69], MEKs 1 and 2 of 44 and 45 kDa, respectively [
70]. The MEK kinase domain contains an ERK-selective binding site near the N-terminus and a nuclear export sequence (NES) that favors its cytosolic localization [
71,
72]. MEK1 is activated by phosphorylation of two activation loop serine residues (S218 and S222, S222 and S226 in MEK2) [
73]. Apart from canonical activation by RAF, MEK can also be activated by other MAP3Ks, including MOS [
74], MAP3K1/MEKK1 [
75], MAP3K8 (aka Tpl2/Cot) [
76], and MAST1 [
77], linking activation to distinct upstream stimuli (
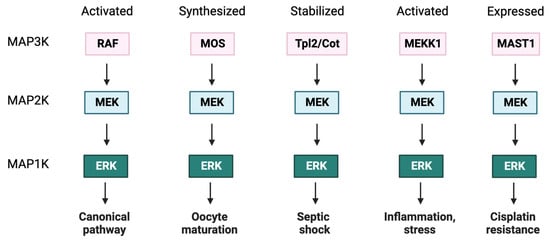
Figure 1).
Figure 1. Activation of MEK by different upstream activators causes diverse outcomes. MEK can be activated by different MAP3Ks in a stimulus- and cell type-specific manner. The canonical pathway involves signal transduction through RAF. The synthesis of MOS induces oocyte maturation by direct phosphorylation of MEK and consequent activation of ERK. Upon stimulation, the kinase Tpl2/Cot is released from a ternary complex and is stabilized by phosphorylation of specific residues. The signaling module Tpl2/Cot-MEK-ERK is important in inflammation and immune response. MEKK1 is an upstream regulator of stress-responsive kinases, and it is also able to phosphorylate MEK1/2 to induce ERK activation in inflammatory settings. MAST1 initial or induced expression leads to cisplatin resistance.
MEK1 is retro-phosphorylated by ERK at T292 [
82]. This phosphorylation event is specific to MEK1 and enhances the accessibility of phosphatases facilitating the dephosphorylation of S218 and S222 in the activation loop [
80]. As MEK1 and MEK2 form heterodimers and this residue is exclusive to MEK1, in the absence of MEK1, MEK2 is not subjected to negative regulation, thus leading to ERK-sustained activation via MEK2 [
83]. As in the case of ERK1/2, MEK1 and MEK2 seem to play non-redundant roles in development [
84,
85]; however, this may be due to a dosage effect [
86].
MEK mutations are rare, found in less than 1% of all human cancers, and not concentrated in a specific region or codon [
63], in contrast to the upstream activators RAF and RAS. Mutations in MEK have been functionally classified into three groups. (1) RAF-independent mutations consist of deletions that result in autophosphorylation in the activation loop, resulting in a potent downstream activation. (2) RAF-regulated alterations lead to some basal activation which is potentiated in the presence of RAF.
2.4. ERK
ERKs contain the usual two-domain protein kinase structure (reviewed in [
93]). The N-terminal domain binds ATP and contains an essential ATP-binding lysine, often referred to as the catalytic lysine. Protein substrates and regulators bind across the C-terminal domain as discussed more fully below. ERKs also include a MAPK insert in the C-terminal domain and other regions that are important for MEK recognition [
94,
95] and an essential C-terminal segment that extends across both domains (
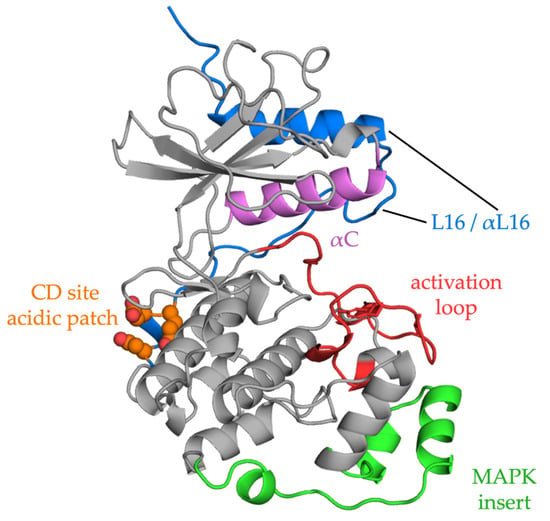
Figure 2).
Figure 2. ERK2 structure. The ribbon diagram shows the two-domain structure of ERK2. Activation of the kinase occurs upon phosphorylation of the TEY motif in the activation loop (red), which lies in front of the active site. Phosphorylation causes a small domain closure, relative to other protein kinases. Reorganization of loop L16 (blue), part of a C-terminal extension that wraps up the back of the kinase, causes repositioning of alpha helix C (αC, magenta), allowing formation of a salt bridge between the glutamate in the helix to the essential lysine in beta strand 3 in the interior of the active site. The acidic residues that make up the CD domain are shown in orange. The FXF binding site lies between the MAPK insert (green) and the kinase core. Interactions through these two docking motifs may provoke conformational changes in ERK2 [
115,
116,
117,
118].
Activation of ERK by MEK results from the phosphorylation of tyrosine followed by threonine in the TEY sequence within the activation loop [
11,
96,
97,
98]. Following phosphorylation by MEK, this C-terminal segment aids in repositioning alpha helix C to achieve an activated ERK conformation [
98]. The combined phosphorylation of both residues increases ERK activity 50,000-fold [
99].
The similarities and overlapping substrate specificities of ERK1 and ERK2 have fueled the long-running debate over their functional specificity [
100,
101,
102,
103]. Genetic studies using knockout mouse models showed that the deletion of either ERK1 or ERK2 led to different phenotypic outcomes. Although loss of either ERK gene impairs neuronal development [
104], ERK1−/− mice are viable and fertile [
105], while whole body ERK2 knockout results in embryonic lethality regardless of ERK1 expression [
106].
N-terminal alanine-rich (NTAR) sequences in ERK1/2 are now known to improve their translation. NTAR sequences ensure precise translation and prevent frameshifts by reducing the speed of elongating ribosomes. Human ERK1/2 possess multiple Ala codons immediately downstream of the AUG codon with an additional in-frame start codon also present.
2.4.1. ERK Substrate Recognition
ERKs can interact with hundreds of substrates in any location in the cell [
119,
120]. In addition to the many phosphorylation sites that have been identified by examining defined targets, many more sites in putative effectors have been predicted by in silico approaches with some validated in vivo [
121,
122]. ERK1/2 show a preference for sites in the consensus phosphorylation sequence, PX(S/T)P, with a proline in the P+1 residue, following the phosphorylation site, and are thus considered proline-directed kinases. Many substrates contain only a minimal (S/T)P sequence.
The huge number of phospho-sites found in proteins [
121] raises the question of how specificity is achieved. While scaffolds can form three-way complexes with enzymes and substrates, docking sites are important MAPK recognition mechanisms [
124,
125]. The best characterized docking sites identified on ERK are the common docking (CD) or D-recruitment site (DRS) and the F-recruitment site (FRS) or docking site for ERK FXF (DEF) (see
Figure 2) [
126]. The CD site, accessible in both inactive and active ERK configurations, binds a basic/hydrophobic motif, typically K/RX
2-4LXL, referred to as a D motif, kinase interaction motif (KIM), or more generally as a short linear motif (SLiM). In addition to many substrates, MEK1/2 also bind ERK1/2 through a CD–D motif interaction, as does the MAP kinase phosphatase DUSP6. The FRS site binds proteins containing FXF motifs, including nuclear pore proteins [
127], and is more accessible after ERK activation [
128,
129].
2.4.2. ERK Localization
ERK1/2 are found in cytoplasmic-, nuclear-, and membrane-associated compartments, and their subcellular distribution defines access to substrates. Localization is dynamic and is influenced by the duration and nature of activating stimulation. Two ERK monomers can associate to form a free dimer in the cytoplasm to activate cytoplasmic substrates. ERK dimers can be assembled on scaffold proteins in specific subcellular localizations where the ERK monomer not associated with the scaffold will phosphorylate specific substrates [
137]. Binding the FG repeat regions of nuclear pore proteins, nucleoporins (Nup) Nup214, Nup153, TPR, and others, allows passive entry of ERK into the nucleus [
138,
139,
140].
While active nuclear import mechanisms have been identified, ERK scaffolding, to retain the kinases in the relevant compartment, and ERK nuclear export through binding to exported partners, appear to dominate processes determining ERK location. As noted earlier, binding to MEK promotes ERK nuclear export and favors cytoplasmic retention. Dual specificity phosphatases also influence ERK location by related mechanisms. The phosphatase DUSP6 is primarily cytoplasmic and binds ERK through a D motif–CD domain interaction; DUSP6 also possesses an NES, promoting ERK nuclear export.
2.4.3. ERK in Cancer
In contrast to upstream cascade components, ERK1/2 are rarely mutated and oncogenic activity has not been unequivocally demonstrated. ERK mutations recognized as of 2020 have been thoroughly reviewed by Smorodinsky-Atias (2020) [
151]. Most of these mutations have been generated in the laboratory based on ERK orthologs, protein structure and two ancestors as follows: one common to ERK1/2/5 with autophosphorylation capabilities and another common to ERK1 and ERK2 that lacks the autophosphorylation ability. A few of them rendered some activation but usually minimal compared to that achieved by MEK [
152].
An ERK mutation with potential oncogenic activities is an intrinsically active mutant ERK1 with the mutation R84S, initially found in a screen for ERK MEK-independent mutants in an ERK yeast ortholog [
154]. This mutant was able to transform NIH3T3 cells [
155] and was activated in developmental assays in a
Drosophila cancer model, especially combined with the sevenmaker mutation [
156]. However, the equivalent mutation in ERK2 did not replicate the transformation outcomes.
According to the COSMIC database, as of September 2023, ERK1 accounts for 0.5% of all the cancer samples analyzed. These mutations can be found throughout the protein with no hotspots identified, most of them were found only in one single sample with a maximum of four cases [
157]. Mutations in ERK2 are slightly more frequent, representing 1.2% of the tested samples. Alterations also spread across the whole protein; however, there is a hotspot in residues 321 and 322 within the common docking domain. In fact, the most common mutation is E322K, which occurs in only 0.1% of all the assessed samples but accounts for 7% of all ERK2 mutations found in cancer samples. Although the majority of the mutations in ERK1 and ERK2 do not follow a tissue distribution pattern, mutations in the CD site of ERK2 are found mainly in squamous cell carcinoma, most frequently in cervix, oral-esophagus, head and neck, lung and oral tissues [
159]. E322K was originally identified in oral squamous cell carcinoma [
160].
3. ERK Functions and Context Specificity
ERK1/2 are critical for integrating extracellular ligand binding cues with intracellular conditions to elicit coordinated cellular responses such as exit from a stem cell state, proliferation, terminal differentiation, and tissue-specific differentiated functions [
165,
166]. As noted above, ERK2 is essential early in embryonic development, and variation in a single allele is associated with developmental and cognitive impairment [
104,
167,
168,
169]. ERK2 can phosphorylate hundreds of downstream targets, including more than half a dozen protein kinase substrates which expand its regulatory reach in many subcellular locations [
122,
141,
170,
171,
172,
173]. The ability to communicate directly and indirectly with such a breadth and distribution of downstream proteins results in both the intrinsic potential of ERK2 to carry out ligand-specific directives and also the malleability of ERK2-elicited responses to context, tightly linked to the cellular proteome expressed at any specific time [
174,
175,
176,
177,
178]. This plasticity also makes cells vulnerable to diseases arising from disturbances in ERK2 regulation. The activities of ERK1/2 must be adjusted up and down during the cell cycle, as development progresses, and in cells of the adult animal. This fluctuation in ERK1/2 activity is required to maintain or exit from a stem cell state, to facilitate terminal differentiation, and to maintain cell identity. ERK1/2 activities are necessary participants that mediate acute endocrine, paracrine, and autocrine stimulation, and adjust outputs due to changes in nutrient availability and other cellular stresses to support the context-specified cellular responses.
3.1. Cell Cycle, Growth, Migration, and Survival
ERK regulates G1 to S phase transition, and its depletion results in cell cycle arrest [
179] (reviewed in [
180]). Cell growth is essentially biomass accumulation due to anabolic growth pathways [
181]. ERK participates in the regulation of several processes implicated in growth for the preparation of cell division (reviewed in [
182]). For instance, ERK stimulates the de novo synthesis of pyrimidine nucleotides to generate DNA and RNA. ERK phosphorylates the CAD protein (carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase), increasing pyrimidine biosynthesis and antagonizing the negative effect of PKA CAD phosphorylation [
183]. Ribosomes are necessary to handle new protein synthesis. ERK stimulates ribosome biogenesis by phosphorylation of RNA polymerase I [
184] and III [
185]. Finally, ERK is involved in protein translation by regulation of mammalian target of rapamycin complex 1 (mTORC1), eukaryotic translation initiation factor 4E (eIF4E) through MNK1/2, and cytoplasmic polyadenylation element binding protein 1 (CPEB1) [
186] (reviewed in [
187]). ERK increases mRNA polyadenylation and translation [
188].
3.2. Context Dependence of ERK Functions in Pancreatic Beta Cells
In pancreatic beta cells, circulating glucose regulates insulin secretion and production. ERK1/2 are essential in nutrient sensing, and their activities rise and fall as a function of glucose concentration over the physiologic range, in parallel with insulin secretion [
203]. Glucose metabolism triggers calcium influx and release from intracellular stores to activate ERK1/2. Calcium influx also activates the calcium-dependent phosphatase calcineurin, which is required for maximal ERK1/2 activation by glucose [
204]. Calcineurin is required for activation of ERK1/2 by other stimuli that induce insulin secretion as well, including depolarization and glucagon-like peptide 1, but not by phorbol ester or insulin. BRAF is also a calcineurin substrate; calcineurin dephosphorylates threonine 401 on BRAF, which is a site of negative feedback phosphorylation by ERK1/2 [
205]. The major calcineurin-dependent event in glucose sensing by ERK1/2 is the activation of BRAF in beta cells [
206].
3.3. ERK in Stemness
The requirement for ERK in stem cells depends on the pluripotency state of the cells. There are two states of pluripotency as follows: naïve, that resembles an early-stage embryo (pre-implantation) and primed stem cells, that evoke a later stage (post-implantation) [
216]. Naïve pluripotent cells require LIF/STAT3 (leukemia inhibitory factor/signal transducer and activator of transcription 3) signaling, whereas primed pluripotency depends on FGF (fibroblast growth factor)/ERK and activin signaling [
217]. ERK activation is dispensable for naïve but essential for primed pluripotency. In the case of mouse embryonic stem cells, the activation of the LIF/STAT3 pathway [
218,
219] and BMP (bone morphogenetic protein) triggering Id (inhibition of differentiation) genes are enough to maintain pluripotency. To avoid differentiation, they are cultured in the presence of LIF [
218] and BMP4 [
220]. In this setting, ERK antagonizes STAT3 signaling promoting differentiation [
221], but BMP4 suppresses ERK signaling to sustain self-renewal through Smad activation and DUSP9 upregulation [
222,
223].
Meanwhile, GSK3 (glycogen synthase kinase-3) inhibits Wnt/β-catenin signaling, which is essential for maintaining self-renewal and inhibiting differentiation in mESCs [
224,
225]. mESCs require MEK and GSK3 inhibitors in the culture medium for stability; this combination is known as 2i-culture [
226,
227]. ERK activation is crucial in mESCs to end self-renewal and initiate differentiation [
228,
229].
Stem cells from mice (mESCs) and human (hESCs) do not behave identically. mESCs are derived from pre-implantation embryos [
231,
232], while hESCs are acquired from post-implantation embryos. Thus, mESCs display naïve pluripotency [
233,
234,
235], whereas hESCs belong to a primed pluripotent state [
236].
3.4. ERK as an Allosteric Regulator
ERK possesses multiple functions independent of its catalytic activity. In addition to the many activity-dependent actions on transcription factors (e.g., Elk1, NeuroD1) noted above, ERK also influences gene expression transcription factors, co-activators, and co-repressors through direct interactions. For instance, ERK acts as a transcriptional repressor for interferon gamma (IFNγ)-induced genes as both ERK2 and C/EBP-β compete to bind GATE element, and only when C/EBP-β is bound are the downstream genes expressed [
244]. ERK directly assembles on a promoter element of the IL4 gene, aiding in recruitment of transcription factors that initiate IL4 gene transcription. Association with the promoter and IL4 transcription induction is necessary for human Th2-cell differentiation [
245]. ERK2 directly binds the MYC promoter and recruits CDK9 which binds and activates RNA polymerase II, resulting in ERK-induced MYC expression independently of ERK activity [
246].
ERK allosterically regulates certain other proteins, notably DUSP6, independent of its kinase activity. A D motif in the non-catalytic amino-terminal domain of DUSP6 binds the ERK CD site. This interaction stabilizes the active conformation of DUSP6 by inducing the closure of the general acid loop in the phosphatase [
248]. Thus, ERK provokes a conformational change in the DUSP6 catalytic domain increasing its phosphatase activity to negatively regulate ERK pathway activation [
249].
4. ERK Regulation
4.1. ERK Negative Feedback Loops
Among its extensive substrates, ERK can bind and phosphorylate upstream members of the cascade to exert a positive or negative impact on the downstream signal. Negative feedback loops downregulate the signaling maintaining pathway balance. In addition to upstream components, ERK can phosphorylate scaffolds and adaptor proteins to rapidly suppress pathway activity, referred to as direct regulation. ERK can also phosphorylate transcription factors responsible for the expression of negative regulators, giving rise to a delayed but long-term response known as indirect regulation (reviewed in [
254]).
ERK can phosphorylate multiple sites on BRAF and CRAF, some preventing their heterodimerization and others the interaction with RAS and localization at the plasma membrane [
255,
256] (
Figure 3). In the case of BRAF, ERK also phosphorylates several residues with a negative outcome [
205]. The conserved motif SPKTP, not present in the other RAF isoforms, is found in the C-terminus of BRAF, and it is also phosphorylated by ERK and implicated in the negative regulation of the pathway [
257]. Three other residues in BRAF, T401, S750 and T753, are phosphorylated by ERK and prevent heterodimerization with CRAF [
205,
258].
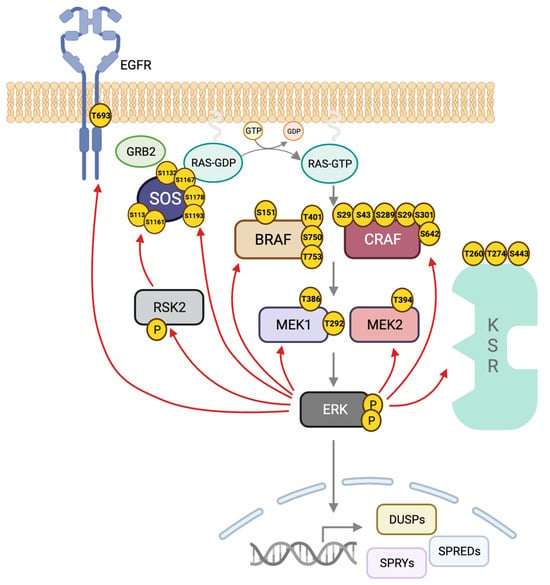
Figure 3. ERK negative feedback loops. ERK phosphorylates MEK1 at T292, thus impeding dimerization with MEK2 and PAK1-mediated phosphorylation of MEK1 in S298. ERK also phosphorylates T386 in MEK1 and the homolog residue in MEK2, T394. In CRAF, there are five ERK phospho-sites throughout the protein as follows: S29 and S43 in the N-terminus, S642 at the C-terminus, and S289, S296 and S301 at the flexible hinge between the regulatory and catalytic domains. The result is impaired localization at the plasma membrane, loss of CRAF–RAS interaction, and enhanced access for PP2A facilitating CRAF dephosphorylation. In BRAF, ERK phosphorylates S151 next to the RAS binding domain (RBD) of RAF, impairing RAS–BRAF interaction. ERK phosphorylates T401, S750 and T753 in BRAF, interfering with BRAF–CRAF dimerization. Pathway activation induces phosphorylation of KSR1 on T260, T274, S320 and S443 by ERK impairing dimerization with RAF and localization in the plasma membrane. ERK directly phosphorylates SOS on S1132, S1167, S1178 and S1193 and indirectly in S1134 and S1161 through RSK2. ERK phosphorylates the EGFR on T693 in the mature form of the receptor.
4.2. ERK Scaffolds
Many actions of the MAPK cascade are targeted to subsets of substrates through association with scaffolding proteins. Nevertheless, the concentrations and affinities of pathway components, which vary widely among cell types [
276], are also important for pathway outputs. Early efforts to quantify relative amounts of these signaling molecules in HeLa and Cos-7 cells suggested that MEK is the most abundant protein of the pathway, followed by ERK, then RAS, and RAF is the least abundant in these cell lines [
277].
The first scaffold characterized in a MAPK cascade was Ste5, identified in the budding yeast Saccharomyces cerevisiae. Ste5 binds Fus3 (a MAPK), Ste7 (a MAP2K) and Ste11 (a MAP3K) forming a multiprotein complex at the tips of mating projections following stimulation with mating pheromones [
294,
295]. Before the discovery of Ste5, it was generally assumed that kinases did not form stable complexes with substrates
Why are scaffolds important? Scaffolds are the least understood components of MAPK pathways, but they connect more directly to functional specificity than the individual cascade kinases themselves. For example, IQGAP1 [
299], paxillin [
300] and GIT1 [
301] ERK scaffolds are responsible for localization of active ERK in focal adhesions and actin filaments.
They incorporate diverse regulatory properties, as detailed below, in addition to serving as cascade assembly sites [
303]. Scaffolds share a number of essential features ensuring enzymes like ERK1/2, which can phosphorylate hundreds of proteins, do so at the right place and time. Scaffold proteins optimize signaling by clustering enzymes and substrates together, increasing their effective concentrations. Scaffolds also position proteins in advantageous orientations, often to facilitate phospho-transfer reactions [
304,
305].
Also central to their function, scaffolds must be at an optimal concentration to produce maximum signal output. With too few scaffold proteins, cascade assembly may fall below the optimum, while too many scaffold proteins will separate the kinase cascade components, yielding unproductive complexes; in the latter case, more is less. This phenomenon has been named “combinatorial inhibition” [
315] and the “prozone effect” [
305,
307,
316,
317]. Ideally, each scaffold should maintain the aforementioned optimum level that results in maximum signal efficiency.
Variations in the expression of a single scaffold have altered total ERK activity to a greater extent than expected by just interfering with a single scaffold [
322,
323,
324,
325,
326,
327]. Furthermore, scaffolds can associate with each other to form macro-complexes. Interactions have been described between MP1 and MORG1 [
328]; IQGAP1 and MP1 [
329]; paxillin and GAB1 [
330]; IQGAP1 and β-arrestin-2 [
331]; and IQGAP1 and KSR1 [
332]. It has been proposed that scaffolds coordinate and cross-talk [
137]. This premise was substantiated a few years later by the functional interaction between KSR1 and IQGAP1. ERK bound to a KSR1 mutant unable to bind MEK can be transactivated from MEK bound to IQGAP1, and this cross-activation mechanism has been named trans-phosphorylation.
4.2.1. KSR1/2
In 1995, KSR1 was simultaneously discovered in
Drosophila virilis/
Drosophila melanogaster [
333] and
Caenorhabditis elegans [
334,
335] by a genetic screen for RAS downstream effectors. The phenotypes of the KSR1 loss-of-function mutants revealed inhibition of RAS signals, and for this reason the protein was named Kinase Suppressor of RAS (KSR) [
333,
334]. KSR was recognized as a mammalian functional equivalent of the yeast MAPK scaffold protein Ste5, even though they do not share sequence homology.
The KSR family is composed of five members, the pseudokinases KSR1 [
333] and KSR2 [
336] and their relatives, the three RAF protein kinases. Despite close sequence similarity between KSR1 and KSR2, these two proteins have distinct functions which are due in part to differences in expression. While KSR1 is widely expressed, KSR2 has a restricted tissue expression pattern, generally with neuroendocrine enrichment. Regarding function, both are scaffolds of the RAS–ERK pathway, but the knockout phenotypes are markedly different. Mice lacking KSR1 show a mild phenotype with disorganized hair follicles but no developmental issues, while RAS-directed oncogenesis is reduced [
323]. On the other hand, KSR2-deficient mice are less fertile and develop metabolic disorders such as obesity, insulin resistance and type 2 diabetes [
337,
338].
Why are KSR1/2 considered pseudokinases? Although mammalian KSR1/2 were initially classified as pseudokinases because they contain arginine in place of the lysine essential for catalytic activity, this has been a point of contention [
348,
349,
350,
351,
352,
353]. Some studies have reported residual kinase activity [
306,
351,
354,
355] not detected by others [
342,
356,
357]. Indirect evidence suggesting catalytic function comes from the findings that ectopic expression of KSR1 or KSR2 is sufficient to induce proliferation in a RAS-independent manner, attributed to heterodimerization with RAF. Still, the exact mechanism by which KSR overexpression leads to ERK activation has not been defined [
358].
KSR1 is usually cytoplasmic and thought to be constitutively bound to MEK [
345,
346,
347,
359]. Upon stimulation and RAS activation, KSR1 rapidly translocates to the plasma membrane where it interacts with a RAF isoform, especially with BRAF [
58]. At the plasma membrane, KSR exhibits selectivity towards defined microdomains, responding preferentially to RAS signals from lipid rafts [
339]. Interaction with the E3 ligase IMP (Impedes Mitogenic signal Propagation) promotes the recruitment of KSR to Triton-resistant structures that sequester KSR1 and block ERK activation [
360,
361]. RAS activation leads to IMP proteasomal degradation, facilitating KSR-mediated ERK activation [
362].
Homo- and heterodimerization of KSR and RAF proteins underscores the remarkable complexity of ERK regulation at the MAP3K level, some of which was originally uncovered through studies of RAF inhibitors. A depth of structural data has revealed multiple dimeric structures of these molecules [
57,
306]. KSR and RAF can form side-to-side heterodimers believed to trigger RAF activation through a nearly identical dimer interface that is conserved across all family members. In the same way, KSR1 can also homodimerize through its C-terminus, forming side-to-side dimers as well [
57,
367]. Apart from KSR–RAF interaction through the kinase domains, selective heterodimerization of RAF with KSR1 occurs through direct contacts between the N-terminal regulatory regions of each protein, including the KSR coiled-coil–sterile alpha motif (CCSAM) [
340].
4.2.2. IQGAP1/2 and 3
IQ motif-containing GTPase Activating Protein (IQGAP) 1 was first identified in 1994 by Weissbach and collaborators in a screen to discover novel matrix metalloproteases [
368]. Nevertheless, it was not until 2004 that IQGAP was classified as a Ras/ERK pathway scaffold [
324]. Mammalian IQGAP1, 2, and 3 are multidomain proteins of ~190 kDa [
369,
370,
371]. IQGAPs exhibit different tissue expression patterns; while IQGAP1 is ubiquitous [
368], IQGAP2 is mainly expressed in the liver and the gastro-intestinal and urogenital track [
372,
373], and IQGAP3 is found mainly in the brain, lung and testes [
374]. IQGAP1 is cytoplasmic and mainly associated with the cytoskeleton, with particular enrichment in cell–cell contacts [
375]. Distributions of IQGAP2 and IQGAP3 are not well described, but they have been noted throughout the cytoplasm and in cell–cell junctions, respectively [
376,
377].
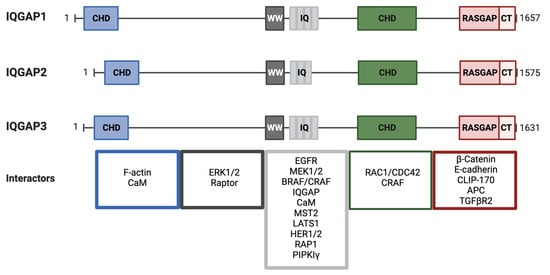
IQGAP isoforms share a similar domain composition (
Figure 5). Abundant protein-interacting motifs make them key players in numerous cellular processes. For example, more than 100 binding partners have been described for IQGAP1 [
379]. At the N-termini of IQGAPs is a calponin homology domain (CHD) that binds cytoskeletal proteins, in particular F-actin [
380]. This domain can also bind calmodulin/Ca
2+ but at a lower affinity than to the IQ domain and in competition with F-actin [
381]. The CHD is followed by a WW domain with two conserved Trp residues (W). WW domains specify proline-rich regions in binding partners. Interestingly, a major IQGAP WW domain-binding partner, ERK, apparently binds through a different mechanism [
324]. Four tandem isoleucine/glutamine-containing (IQ) motifs are multipurpose domains that bind partners including calmodulin [
381,
382], the EGF receptor [
383], MEK [
384], BRAF [
385] and the small GTPase RAP1 [
386]. IQ domains are also important as they mediate the formation of IQGAP dimers and oligomers [
387]. IQ domains participate in the interaction with phosphoinositide signaling elements such as phosphatidylinositol phosphate kinase type Iγ (PIPKIγ) at the leading edge. PIPKIγ binding favors the IQGAP open conformation to recruit the actin polymerization machinery and participate in cell motility [
388].
Figure 5. Schematic of IQGAP1/2 and 3 domain organization and interactors. IQGAPs are divided into five conserved domains, from N- to C-terminus. CHD, calponin homology domain; WW domain, a poly-proline protein–protein domain with two conserved Trp (W) residues; IQ domain, with four repeats (three for IQGAP2) of isoleucine- and glutamine-containing motifs; GRD, GAP-related domain; and RGCT, RAS GAP C-terminus domain. Some representative binding partners for each domain are organized and displayed in the color-coded boxes.
Despite the long list of interactors, a major role of IQGAP1 is as a scaffold for the RAS/ERK pathway [
324]. IQGAP1 binds both MEK and ERK [
384] and can also interact with BRAF and modulate its functions. Generally, IQGAP1–BRAF association is decreased by Ca
2+/calmodulin binding to IQGAP1, as this interaction provokes a conformational change [
393]. IQGAP1-bound BRAF has higher kinase activity than free BRAF; it is unclear whether binding to IQGAP1 promotes high BRAF activity or whether highly activated BRAF preferentially binds IQGAP [
393]. EGF stimulates IQGAP1 binding to MEK1 and decreases binding to MEK2. It has been suggested that MEK1 binding to IQGAP1 promotes proliferation, whereas binding to MEK2 induces differentiation [
394]. In contrast to MEK and BRAF, ERK1/2 constitutively bind IQGAP [
324].
Under the control of RAC1/Cdc42 and antagonizing Ca
2+/calmodulin signals, IQGAP1 participates in cell migration through focal adhesions and cytoskeleton dynamics by mediating co-localization of N-WASP with the Arp2/3 complex in lamellipodia which assemble to catalyze actin polymerization [
398,
399,
400]. Stimuli that promote the formation of focal adhesions, such as PDGF, stimulate the assembly of IQGAP1 complexes with the focal adhesion proteins vinculin and paxillin [
401]. During migration, a coordinated assembly and disassembly of focal adhesions is necessary. MP1 (MEK partner 1 aka LAMTOR3) plays a role in this process by anchoring MEK to late endosomes via p14 (LAMTOR2) [
327]. Knock down of MP1 impairs migration due to the formation of abnormal focal adhesions that accumulate IQGAP1, suggesting that the interaction between IQGAP1, MP1 and the ERK cascade is crucial in this process [
329]. IQGAP1 at the leading edge in migrating cells promotes migration in a RAC1- and Cdc42-dependent fashion.
IQGAP1, like KSR1, is not required for embryonic development. Mice lacking IQGAP1 are viable and fertile with gastric hyperplasia as a mild phenotype [
408]. However, these mice are resistant to developing tumors induced by oncogenic H-RAS [
325]. In the case of IQGAP2 knockout mice, they develop age-dependent hepatocellular carcinoma (HCC) with increased IQGAP1 expression [
409]. These two phenotypes align with IQGAP1 pro-tumorigenic and IQGAP2 anti-tumorigenic roles.
4.2.3. HPIP
HPIP (hematopoietic PBX (pre-B cell leukemia homeobox)-interacting protein or pre-B cell leukemia transcription factor-interacting protein (PBXIP1)) is a 731-amino acid protein that was discovered in 2000 in a yeast two-hybrid screen using PBX1a as bait. HPIP interacts with PBX transcription factors, negatively regulating PBX1-mediated hematopoiesis [
417]. Although HPIP is predominantly cytoplasmic, specifically localizing on microtubules, this protein contains both a nuclear localization signal (NLS) and a nuclear export signal (NES) [
418].
It was not until ten years after its discovery that HPIP was recognized as an ERK scaffold. HPIP associates with the estrogen receptor (ERα), Src and PI3K, which, in turn, activate ERK1/2 and AKT signaling pathways [
419]. Thus, HPIP’s function as a scaffold protein is not exclusive to the RAS–ERK pathway. HPIP participates in signal transduction via transcription factors, C/EBPα and GATA1, ERα and ERβ, LEF1/ β-catenin and p53 as well as through tyrosine and serine/threonine protein kinases, FAK/Src, and TBK1 and CK1α, and it has also been implicated in the regulation by TGF-β. This great variety of interactors connect HPIP with processes including hematopoiesis, cell proliferation and tumorigenesis, germ cell proliferation, EMT, renal fibrosis, cell migration and invasion, and osteoarthritis. HPIP is widely expressed in different tissues, and it is considered a proto-oncogene since it is overexpressed in more than 15 types of cancer [
420].
5. Pathway Inhibitors
5.1. Classical Inhibitors
Many classical inhibitors for the different members of the cascade have been developed. Many of them are in clinical trials but only a few have received FDA approval, mostly those targeting RAF and MEK. Remarkably, two covalent inhibitors, sotorasib and adragasib, have been FDA-approved in the last couple of years to treat KRAS
G12C lung cancers (reviewed in [
487]), and several promising clinical trials are currently in progress [
30]. As of today, no ERK inhibitors are in clinical use, although several are in clinical trials [
488].
In contrast to many other kinases, ERK2 undergoes relatively small conformational changes in the active site upon activation. Solution measurements have found evidence for phosphorylated ERK2 (2P-ERK2) in two conformational states, R and L, which interconvert on a millisecond time scale [
490]. Surprisingly, the L state resembles the active site of unphosphorylated ERK (0P-ERK2). A recent study found ERK inhibitors that interact with both states and ones that selectively interact with the R or L states. Inhibitors were noted that could shift the equilibrium towards the R or L states. As some ERK inhibitors have been shown to affect specific protein–protein interactions, this study provides new insights for inhibitor design [
491].
5.2. PROTAC Technology Applied to the ERK Pathway
As its name implies, Targeted Protein Degradation (TPD) covers several methods to induce the selective degradation of a specific molecule either via the proteasome, lysosome or autophagy system. One of these methods using the proteasome degradation system is PROTAC (Proteolysis-Targeting Chimera) [
514]
, initially developed in 2001 [
515,
516,
517,
518]. The strategy involves recruitment of an E3 ligase, usually VHL (von Hippel-Lindau VHL) or CRBN (Cereblon), in current versions of PROTACs, to the target protein to tag it for degradation. Two ligands, one that binds the target protein and a second that recruits an E3 ligase, are connected through a chemical linker. The target protein will be ubiquitinylated by the E3 and sent to the proteasome resulting in its near elimination [
519]. Advantages of protein degradation versus classical small molecule inhibitors are many. PROTACs can reach the undruggable proteome as long as a suitable target ligand is available. Degradation prevents target protein accumulation, thus avoiding compensatory upregulation and drug resistance. PROTACs overcome the generation of resistant mutations under selective pressures or even the emergence of non-enzymatic functions of the targeted proteins, which might provoke a signaling rebound.
The major limitation of PROTACs continues to be poor oral bioavailability and low cell permeability due to their large sizes [
523]. For this reason, it was not until 2019 that the first PROTAC progressed to clinical trials [
524]. Although none are employed in the RAS–ERK pathway, a few FDA-approved therapies use degraders [
525].
The first attempt to generate a PROTAC against KRASG12C was based on the inhibitor ARS1620, and a thalidomide analog, pomalidomide, an FDA-approved drug with high affinity for the ubiquitin ligase CRBN. The compound was named XY-4-88, but failed to induce endogenous KRASG12C degradation in cancer cells [
526]. A parallel study generated LC-2, the first successful PROTAC against KRASG12C, which used MRTX849 as the parental inhibitor [
527].
The first series of compounds acting as PROTACs for BRAF were created based on the inhibitor rigosertib, which binds to RAF in the RAS binding domain (RBD) and should, therefore, bind RAF mutants as activating mutations are in the kinase domain [
529], although this was not tested. P4B was the first effective PROTAC with selectivity for the BRAF mutant V600E. The ligand was based on the inhibitor BI882370 and provides degradative efficacy in the nanomolar range [
530].
The first-in-class highly selective degrader of MEK1 and MEK2 was the PD0325901-based compound MS432. It was effective in colorectal cancer and melanoma cell proliferation, with good bioavailability in mice [
534]. A few PROTAC compounds against MEK1 and MEK2 were developed based on allosteric MEK inhibitors, such as PD0325901 and refametinib, using VHL as the E3 ligase. They were assessed in cell proliferation and pERK levels to compare the compound with the parental inhibitor. Although these compounds had modest degradation efficiencies, two of them were able to completely suppress proliferation in melanoma cells [
535]. Following the development of the first-in-class MEK degrader, the Lin laboratory also characterized three more, MS928, MS934 and MS910. Compared with previously reported VHL-recruiting degraders, these new compounds were more potent in preserving the high selectivity. Of these three degraders, MS934 appeared as the best MEK1/2 degrader for in vivo studies to date.
5.3. Molecular Glues Targeting MEK
The natural products cyclosporin A and FK506 were among the earliest described as molecular glues by Schreiber in 1992 for their ability to promote interactions between two molecules that do not otherwise bind. Following this molecular glue concept, Simonetta (2019) [
543] set out to develop a compound that would enhance a protein–protein interaction between a transcription factor and the E3 that normally degrades it. Subsequently, more molecular glues have been identified by high-throughput chemical screens and target validation [
544,
545].
Because molecular glues are small molecules, unlike PROTACs, they should have superior membrane penetrability and oral bioavailability. On the other hand, PROTACs display a higher adaptability to target a protein of interest, while the original molecular glues had unknown ligands [
523,
546].
5.4. Optically Activated MEK Inhibitors
Light-dependent pharmacological approaches, specifically photo-decaging, have been proposed to prevent MEKi toxicities. The strategy consists of generating an inactive locked MEK inhibitor precursor, by linking a photo-cleavable protecting group (the ‘cage’) to the small molecule inhibitor. This drug protector is irreversibly cleaved from the MEK inhibitor by exposure to light, and then the inhibitor will bind MEK where it has been uncaged. This cutting-edge strategy has been carried out with the potent allosteric inhibitor PD0325901, which was previously discontinued in clinical trials due to its harmful effects which are presumably on-target. The optically activatable MEK1/2 inhibitors, named opti-MEKi, have shown promising efficacy in vivo in a melanoma xenograft zebrafish model [550]. Combinations of these approaches, e.g., photo-switchable linkers or light-activated degraders, are also in development [551].