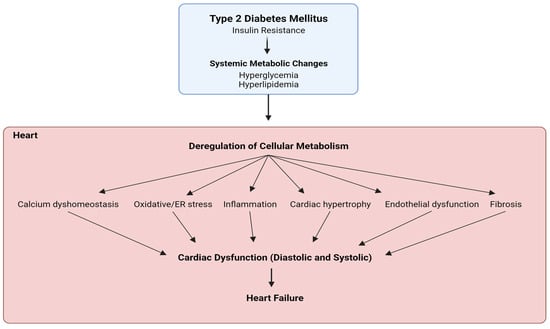
Diabetic cardiomyopathy is a critical diabetes-mediated co-morbidity characterized by cardiac dysfunction and heart failure, without predisposing hypertensive or atherosclerotic conditions. Metabolic insulin resistance, promoting hyperglycemia and hyperlipidemia, is the primary cause of diabetes-related disorders, but ambiguous tissue-specific insulin sensitivity has shed light on the importance of identifying a unified target paradigm for both the glycemic and non-glycemic context of type 2 diabetes (T2D). Several studies have indicated hyperactivation of the mammalian target of rapamycin (mTOR), specifically complex 1 (mTORC1), as a critical mediator of T2D pathophysiology by promoting insulin resistance, hyperlipidemia, inflammation, vasoconstriction, and stress. Moreover, mTORC1 inhibitors like rapamycin and their analogs have shown significant benefits in diabetes and related cardiac dysfunction. Recently, FDA-approved anti-hyperglycemic sodium–glucose co-transporter 2 inhibitors (SGLT2is) have gained therapeutic popularity for T2D and diabetic cardiomyopathy, even acknowledging the absence of SGLT2 channels in the heart. Recent studies have proposed SGLT2-independent drug mechanisms to ascertain their cardioprotective benefits by regulating sodium homeostasis and mimicking energy deprivation.
- diabetes
- diabetic cardiomyopathy
- SGLT2i
- mTORC1/2
- AMPK
1. Diabetic Cardiomyopathy
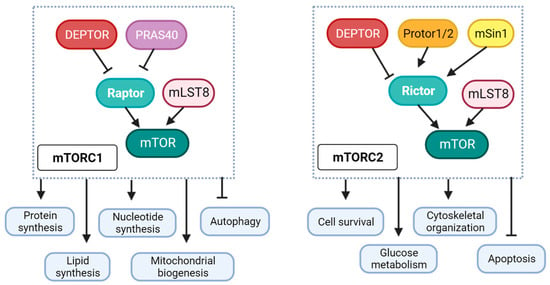
2. Differential Role of mTORC1 and mTORC2 in Diabetic Cardiomyopathy
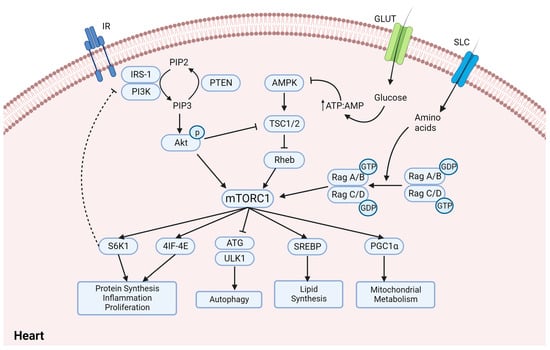
3. Sodium and Glucose Co-Transporter Inhibitors (SGLT2is)—Do They Regulate mTORC1 in Diabetic Cardiomyopathy?
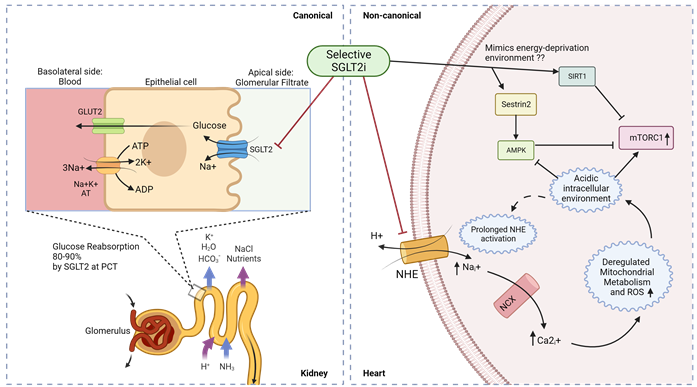
Figure 4. Canonical and non-canonical (cardiac) mechanism for SGLT2i function in diabetic cardiomyopathy. Luminal glucose in the renal glomerular filtrate is reabsorbed (80–90%) into the proximal tubular epithelial cells via SGLT2 in the S1 segment of proximal convoluted tubule (PCT) of nephrons, and subsequently transported to the basolateral interstitial fluid through GLUT2. In diabetic cardiomyopathy therapy, SGLT2is act in a canonical manner by preventing renal glucose reabsorption to reduce overall hyperglycemia but studies indicate a non-canonical mechanism of SGLT2is in the heart. Owing to the absence of SGLT2 channels in the heart, selective SGLT2is can act on prolonged activated NHE to reduce intracellular sodium ions and subsequently promote an alkaline intracellular pH, which has been reported to activate AMPK and inhibit mTORC1. SGLT2 inhibitors have also been reported to mimic an energy-deprived environment that might promote Sestrin2-mediated AMPK and SIRT1 activation, leading to mTORC1 inactivation and cardiovascular benefits. PCT—proximal convoluted tubule; ; NHE—sodium–hydrogen exchanger; NCX—sodium–calcium exchanger.
4. Conclusions
This entry is adapted from the peer-reviewed paper 10.3390/ijms242015078
References
- IDF Diabetes Atlas 2021|IDF Diabetes Atlas. Available online: https://diabetesatlas.org/atlas/tenth-edition/ (accessed on 21 April 2023).
- Kautzky-Willer, A.; Leutner, M.; Harreiter, J. Sex Differences in Type 2 Diabetes. Diabetologia 2023, 66, 986–1002.
- Francisco, P.M.S.B.; de Assumpção, D.; Bacurau, A.G.d.M.; da Silva, D.S.M.; Yassuda, M.S.; Borim, F.S.A. Diabetes Mellitus in Older Adults, Prevalence and Incidence: Results of the FIBRA Study. Rev. Bras. Geriatr. Gerontol. 2022, 25, e210203.
- Tancredi, M.; Rosengren, A.; Svensson, A.-M.; Kosiborod, M.; Pivodic, A.; Gudbjörnsdottir, S.; Wedel, H.; Clements, M.; Dahlqvist, S.; Lind, M. Excess Mortality among Persons with Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 1720–1732.
- Kannel, W.B.; McGee, D.L. Diabetes and Cardiovascular Disease. The Framingham Study. JAMA 1979, 241, 2035–2038.
- Lundbaek, K. Diabetic Angiopathy: A Specific Vascular Disease. Lancet 1954, 266, 377–379.
- Rubler, S.; Dlugash, J.; Yuceoglu, Y.Z.; Kumral, T.; Branwood, A.W.; Grishman, A. New Type of Cardiomyopathy Associated with Diabetic Glomerulosclerosis. Am. J. Cardiol. 1972, 30, 595–602.
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2013, 62, e147–e239.
- Authors/Task Force Members; Rydén, L.; Grant, P.J.; Anker, S.D.; Berne, C.; Cosentino, F.; Danchin, N.; Deaton, C.; Escaned, J.; Hammes, H.-P.; et al. ESC Guidelines on Diabetes, Pre-Diabetes, and Cardiovascular Diseases Developed in Collaboration with the EASD: The Task Force on Diabetes, Pre-Diabetes, and Cardiovascular Diseases of the European Society of Cardiology (ESC) and Developed in Collaboration with the European Association for the Study of Diabetes (EASD). Eur. Heart J. 2013, 34, 3035–3087.
- Goyal, R.; Jialal, I. Type 2 Diabetes. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023.
- Tan, Y.; Zhang, Z.; Zheng, C.; Wintergerst, K.A.; Keller, B.B.; Cai, L. Mechanisms of Diabetic Cardiomyopathy and Potential Therapeutic Strategies: Preclinical and Clinical Evidence. Nat. Rev. Cardiol. 2020, 17, 585–607.
- Taylor, R. Insulin Resistance and Type 2 Diabetes. Diabetes 2012, 61, 778–779.
- Riehle, C.; Bauersachs, J. Of Mice and Men: Models and Mechanisms of Diabetic Cardiomyopathy. Basic Res. Cardiol. 2018, 114, 2.
- Hölscher, M.E.; Bode, C.; Bugger, H. Diabetic Cardiomyopathy: Does the Type of Diabetes Matter? Int. J. Mol. Sci. 2016, 17, 2136.
- Lebeche, D.; Davidoff, A.J.; Hajjar, R.J. Interplay between Impaired Calcium Regulation and Insulin Signaling Abnormalities in Diabetic Cardiomyopathy. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 715–724.
- Buchanan, J.; Mazumder, P.K.; Hu, P.; Chakrabarti, G.; Roberts, M.W.; Yun, U.J.; Cooksey, R.C.; Litwin, S.E.; Abel, E.D. Reduced Cardiac Efficiency and Altered Substrate Metabolism Precedes the Onset of Hyperglycemia and Contractile Dysfunction in Two Mouse Models of Insulin Resistance and Obesity. Endocrinology 2005, 146, 5341–5349.
- Bonen, A.; Jain, S.S.; Snook, L.A.; Han, X.-X.; Yoshida, Y.; Buddo, K.H.; Lally, J.S.; Pask, E.D.; Paglialunga, S.; Beaudoin, M.-S.; et al. Extremely Rapid Increase in Fatty Acid Transport and Intramyocellular Lipid Accumulation but Markedly Delayed Insulin Resistance after High Fat Feeding in Rats. Diabetologia 2015, 58, 2381–2391.
- Jia, G.; Whaley-Connell, A.; Sowers, J.R. Diabetic Cardiomyopathy: A Hyperglycaemia- and Insulin-Resistance-Induced Heart Disease. Diabetologia 2018, 61, 21–28.
- Nesti, L.; Natali, A. Metformin Effects on the Heart and the Cardiovascular System: A Review of Experimental and Clinical Data. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 657–669.
- Koka, S.; Das, A.; Salloum, F.N.; Kukreja, R.C. Phosphodiesterase-5 Inhibitor Tadalafil Attenuates Oxidative Stress and Protects against Myocardial Ischemia/Reperfusion Injury in Type 2 Diabetic Mice. Free Radic. Biol. Med. 2013, 60, 80–88.
- Pavillard, L.E.; Cañadas-Lozano, D.; Alcocer-Gómez, E.; Marín-Aguilar, F.; Pereira, S.; Robertson, A.A.B.; Muntané, J.; Ryffel, B.; Cooper, M.A.; Quiles, J.L.; et al. NLRP3-Inflammasome Inhibition Prevents High Fat and High Sugar Diets-Induced Heart Damage through Autophagy Induction. Oncotarget 2017, 8, 99740–99756.
- Gu, J.; Cheng, Y.; Wu, H.; Kong, L.; Wang, S.; Xu, Z.; Zhang, Z.; Tan, Y.; Keller, B.B.; Zhou, H.; et al. Metallothionein Is Downstream of Nrf2 and Partially Mediates Sulforaphane Prevention of Diabetic Cardiomyopathy. Diabetes 2017, 66, 529–542.
- Wu, L.; Wang, K.; Wang, W.; Wen, Z.; Wang, P.; Liu, L.; Wang, D.W. Glucagon-like Peptide-1 Ameliorates Cardiac Lipotoxicity in Diabetic Cardiomyopathy via the PPARα Pathway. Aging Cell 2018, 17, e12763.
- Brown, M.S.; Goldstein, J.L. Selective versus Total Insulin Resistance: A Pathogenic Paradox. Cell Metab. 2008, 7, 95–96.
- da Silva, A.A.; do Carmo, J.M.; Li, X.; Wang, Z.; Mouton, A.J.; Hall, J.E. Role of Hyperinsulinemia and Insulin Resistance in Hypertension: Metabolic Syndrome Revisited. Can. J. Cardiol. 2020, 36, 671–682.
- Gallagher, E.J.; LeRoith, D. Hyperinsulinaemia in Cancer. Nat. Rev. Cancer 2020, 20, 629–644.
- Kolb, H.; Stumvoll, M.; Kramer, W.; Kempf, K.; Martin, S. Insulin Translates Unfavourable Lifestyle into Obesity. BMC Med. 2018, 16, 232.
- Herman, M.E.; O’Keefe, J.H.; Bell, D.S.H.; Schwartz, S.S. Insulin Therapy Increases Cardiovascular Risk in Type 2 Diabetes. Prog. Cardiovasc. Dis. 2017, 60, 422–434.
- Einarson, T.R.; Acs, A.; Ludwig, C.; Panton, U.H. Prevalence of Cardiovascular Disease in Type 2 Diabetes: A Systematic Literature Review of Scientific Evidence from across the World in 2007–2017. Cardiovasc. Diabetol. 2018, 17, 83.
- Yoneyama, Y.; Inamitsu, T.; Chida, K.; Iemura, S.-I.; Natsume, T.; Maeda, T.; Hakuno, F.; Takahashi, S.-I. Serine Phosphorylation by mTORC1 Promotes IRS-1 Degradation through SCFβ-TRCP E3 Ubiquitin Ligase. iScience 2018, 5, 1–18.
- Hsu, P.P.; Kang, S.A.; Rameseder, J.; Zhang, Y.; Ottina, K.A.; Lim, D.; Peterson, T.R.; Choi, Y.; Gray, N.S.; Yaffe, M.B.; et al. The mTOR-Regulated Phosphoproteome Reveals a Mechanism of mTORC1-Mediated Inhibition of Growth Factor Signaling. Science 2011, 332, 1317–1322.
- Um, S.H.; Frigerio, F.; Watanabe, M.; Picard, F.; Joaquin, M.; Sticker, M.; Fumagalli, S.; Allegrini, P.R.; Kozma, S.C.; Auwerx, J.; et al. Absence of S6K1 Protects against Age- and Diet-Induced Obesity While Enhancing Insulin Sensitivity. Nature 2004, 431, 200–205.
- Malhowski, A.J.; Hira, H.; Bashiruddin, S.; Warburton, R.; Goto, J.; Robert, B.; Kwiatkowski, D.J.; Finlay, G.A. Smooth Muscle Protein-22-Mediated Deletion of Tsc1 Results in Cardiac Hypertrophy That Is mTORC1-Mediated and Reversed by Rapamycin. Hum. Mol. Genet. 2011, 20, 1290–1305.
- Völkers, M.; Toko, H.; Doroudgar, S.; Din, S.; Quijada, P.; Joyo, A.Y.; Ornelas, L.; Joyo, E.; Thuerauf, D.J.; Konstandin, M.H.; et al. Pathological Hypertrophy Amelioration by PRAS40-Mediated Inhibition of mTORC1. Proc. Natl. Acad. Sci. USA 2013, 110, 12661–12666.
- Sciarretta, S.; Forte, M.; Frati, G.; Sadoshima, J. New Insights Into the Role of mTOR Signaling in the Cardiovascular System. Circ. Res. 2018, 122, 489–505.
- Bell, D.S.H. Heart Failure: The Frequent, Forgotten, and Often Fatal Complication of Diabetes. Diabetes Care 2003, 26, 2433–2441.
- Sciarretta, S.; Zhai, P.; Shao, D.; Maejima, Y.; Robbins, J.; Volpe, M.; Condorelli, G.; Sadoshima, J. Rheb Is a Critical Regulator of Autophagy during Myocardial Ischemia: Pathophysiological Implications in Obesity and Metabolic Syndrome. Circulation 2012, 125, 1134–1146.
- Despa, S.; Islam, M.A.; Weber, C.R.; Pogwizd, S.M.; Bers, D.M. Intracellular Na+ Concentration Is Elevated in Heart Failure but Na/K Pump Function Is Unchanged. Circulation 2002, 105, 2543–2548.
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638.
- Luo, M.; Anderson, M.E. Mechanisms of Altered Ca2+ Handling in Heart Failure. Circ. Res. 2013, 113, 690–708.
- Ogawa, A.; Firth, A.L.; Smith, K.A.; Maliakal, M.V.; Yuan, J.X.-J. PDGF Enhances Store-Operated Ca2+ Entry by Upregulating STIM1/Orai1 via Activation of Akt/mTOR in Human Pulmonary Arterial Smooth Muscle Cells. Am. J. Physiol. Cell Physiol. 2012, 302, C405–C411.
- Cang, C.; Zhou, Y.; Navarro, B.; Seo, Y.-J.; Aranda, K.; Shi, L.; Battaglia-Hsu, S.; Nissim, I.; Clapham, D.E.; Ren, D. mTOR Regulates Lysosomal ATP-Sensitive Two-Pore Na+ Channels to Adapt to Metabolic State. Cell 2013, 152, 778–790.
- Hisatsune, C.; Shimada, T.; Miyamoto, A.; Lee, A.; Yamagata, K. Tuberous Sclerosis Complex (TSC) Inactivation Increases Neuronal Network Activity by Enhancing Ca2+ Influx via L-Type Ca2+ Channels. J. Neurosci. 2021, 41, 8134–8149.
- Amemiya, Y.; Maki, M.; Shibata, H.; Takahara, T. New Insights into the Regulation of mTOR Signaling via Ca2+-Binding Proteins. Int. J. Mol. Sci. 2023, 24, 3923.
- Sanlialp, A.; Schumacher, D.; Kiper, L.; Varma, E.; Riechert, E.; Ho, T.C.; Hofmann, C.; Kmietczyk, V.; Zimmermann, F.; Dlugosz, S.; et al. Saraf-Dependent Activation of mTORC1 Regulates Cardiac Growth. J. Mol. Cell Cardiol. 2020, 141, 30–42.
- Ogunbayo, O.A.; Duan, J.; Xiong, J.; Wang, Q.; Feng, X.; Ma, J.; Zhu, M.X.; Evans, A.M. mTORC1 Controls Lysosomal Ca2+ Release through the Two-Pore Channel TPC2. Sci. Signal 2018, 11, eaao5775.
- Janse, M.J. Electrophysiological Changes in Heart Failure and Their Relationship to Arrhythmogenesis. Cardiovasc. Res. 2004, 61, 208–217.
- Liu, C.; Liu, E.; Luo, T.; Zhang, W.; He, R. Opening of the Inward Rectifier Potassium Channel Alleviates Maladaptive Tissue Repair Following Myocardial Infarction. Acta Biochim. Biophys. Sin. 2016, 48, 687–695.
- Liu, Q.-H.; Zhang, L.-J.; Wang, J.; Wu, B.-W.; Cao, J.-M. Cardioprotection of an IK1 Channel Agonist on L-Thyroxine Induced Rat Ventricular Remodeling. Am. J. Transl. Res. 2021, 13, 8683–8696.
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-Activated Protein Kinase in Mechanism of Metformin Action. J. Clin. Investig. 2001, 108, 1167–1174.
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brûlé, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J.; et al. Metformin, Independent of AMPK, Inhibits mTORC1 in a Rag GTPase-Dependent Manner. Cell Metab. 2010, 11, 390–401.
- Das, A.; Durrant, D.; Koka, S.; Salloum, F.N.; Xi, L.; Kukreja, R.C. Mammalian Target of Rapamycin (mTOR) Inhibition with Rapamycin Improves Cardiac Function in Type 2 Diabetic Mice. J. Biol. Chem. 2014, 289, 4145–4160.
- Reifsnyder, P.C.; Flurkey, K.; Te, A.; Harrison, D.E. Rapamycin Treatment Benefits Glucose Metabolism in Mouse Models of Type 2 Diabetes. Aging 2016, 8, 3120–3130.
- McMullen, J.R.; Sherwood, M.C.; Tarnavski, O.; Zhang, L.; Dorfman, A.L.; Shioi, T.; Izumo, S. Inhibition of mTOR Signaling With Rapamycin Regresses Established Cardiac Hypertrophy Induced by Pressure Overload. Circulation 2004, 109, 3050–3055.
- Houde, V.P.; Brûlé, S.; Festuccia, W.T.; Blanchard, P.-G.; Bellmann, K.; Deshaies, Y.; Marette, A. Chronic Rapamycin Treatment Causes Glucose Intolerance and Hyperlipidemia by Upregulating Hepatic Gluconeogenesis and Impairing Lipid Deposition in Adipose Tissue. Diabetes 2010, 59, 1338–1348.
- Salmon, A.B. About-Face on the Metabolic Side Effects of Rapamycin. Oncotarget 2015, 6, 2585–2586.
- American Diabetes Association. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes—2021. Diabetes Care 2020, 44, S111–S124.
- Yang, F.; Qin, Y.; Wang, Y.; Meng, S.; Xian, H.; Che, H.; Lv, J.; Li, Y.; Yu, Y.; Bai, Y.; et al. Metformin Inhibits the NLRP3 Inflammasome via AMPK/mTOR-Dependent Effects in Diabetic Cardiomyopathy. Int. J. Biol. Sci. 2019, 15, 1010–1019.
- Baker, C.; Retzik-Stahr, C.; Singh, V.; Plomondon, R.; Anderson, V.; Rasouli, N. Should Metformin Remain the First-Line Therapy for Treatment of Type 2 Diabetes? Ther. Adv. Endocrinol. Metab. 2021, 12, 2042018820980225.
- Mao, Z.; Zhang, W. Role of mTOR in Glucose and Lipid Metabolism. Int. J. Mol. Sci. 2018, 19, 2043.
- Koepsell, H. The Na+-D-Glucose Cotransporters SGLT1 and SGLT2 Are Targets for the Treatment of Diabetes and Cancer. Pharmacol. Ther. 2017, 170, 148–165.
- Keller, D.M.; Ahmed, N.; Tariq, H.; Walgamage, M.; Walgamage, T.; Mohammed, A.; Chou, J.T.-T.; Kałużna-Oleksy, M.; Lesiak, M.; Straburzyńska-Migaj, E. SGLT2 Inhibitors in Type 2 Diabetes Mellitus and Heart Failure—A Concise Review. J. Clin. Med. 2022, 11, 1470.
- Bakris, G.L.; Fonseca, V.A.; Sharma, K.; Wright, E.M. Renal Sodium–Glucose Transport: Role in Diabetes Mellitus and Potential Clinical Implications. Kidney Int. 2009, 75, 1272–1277.
- Wilcox, C.S. Antihypertensive and Renal Mechanisms of SGLT2 (Sodium-Glucose Linked Transporter 2) Inhibitors. Hypertension 2020, 75, 894–901.
- Kosiborod, M.; Lam, C.S.P.; Kohsaka, S.; Kim, D.J.; Karasik, A.; Shaw, J.; Tangri, N.; Goh, S.-Y.; Thuresson, M.; Chen, H.; et al. Cardiovascular Events Associated With SGLT-2 Inhibitors Versus Other Glucose-Lowering Drugs: The CVD-REAL 2 Study. J. Am. Coll. Cardiol. 2018, 71, 2628–2639.
- Lopaschuk, G.D.; Verma, S. Mechanisms of Cardiovascular Benefits of Sodium Glucose Co-Transporter 2 (SGLT2) Inhibitors: A State-of-the-Art Review. JACC Basic Transl. Sci. 2020, 5, 632–644.
- Huang, K.; Luo, X.; Liao, B.; Li, G.; Feng, J. Insights into SGLT2 Inhibitor Treatment of Diabetic Cardiomyopathy: Focus on the Mechanisms. Cardiovasc. Diabetol. 2023, 22, 86.
- Banerjee, S.K.; McGaffin, K.R.; Pastor-Soler, N.M.; Ahmad, F. SGLT1 Is a Novel Cardiac Glucose Transporter That Is Perturbed in Disease States. Cardiovasc. Res. 2009, 84, 111–118.
- Li, Y.; Xu, G. Sodium Glucose Cotransporter 1 (SGLT1) Inhibitors in Cardiovascular Protection: Mechanism Progresses and Challenges. Pharmacol. Res. 2022, 176, 106049.
- Zhao, M.; Li, N.; Zhou, H. SGLT1: A Potential Drug Target for Cardiovascular Disease. DDDT 2023, 17, 2011–2023.
- Filippatos, T.D.; Liontos, A.; Papakitsou, I.; Elisaf, M.S. SGLT2 Inhibitors and Cardioprotection: A Matter of Debate and Multiple Hypotheses. Postgrad. Med. 2019, 131, 82–88.
- Trum, M.; Riechel, J.; Wagner, S. Cardioprotection by SGLT2 Inhibitors—Does It All Come Down to Na+? Int. J. Mol. Sci. 2021, 22, 7976.
- Ion Channels in the Heart—Bartos—Major Reference Works—Wiley Online Library. Available online: https://onlinelibrary.wiley.com/doi/10.1002/cphy.c140069 (accessed on 24 September 2023).
- Bertero, E.; Prates Roma, L.; Ameri, P.; Maack, C. Cardiac Effects of SGLT2 Inhibitors: The Sodium Hypothesis. Cardiovasc. Res. 2018, 114, 12–18.
- Bell, R.M.; Yellon, D.M. SGLT2 Inhibitors: Hypotheses on the Mechanism of Cardiovascular Protection. Lancet Diabetes Endocrinol. 2018, 6, 435–437.
- Mascolo, A.; Di Napoli, R.; Balzano, N.; Cappetta, D.; Urbanek, K.; De Angelis, A.; Scisciola, L.; Di Meo, I.; Sullo, M.G.; Rafaniello, C.; et al. Safety Profile of Sodium Glucose Co-Transporter 2 (SGLT2) Inhibitors: A Brief Summary. Front. Cardiovasc. Med. 2022, 9, 1010693.
- Chen, S.; Coronel, R.; Hollmann, M.W.; Weber, N.C.; Zuurbier, C.J. Direct Cardiac Effects of SGLT2 Inhibitors. Cardiovasc. Diabetol. 2022, 21, 45.
- Baartscheer, A.; Schumacher, C.A.; van Borren, M.M.G.J.; Belterman, C.N.W.; Coronel, R.; Opthof, T.; Fiolet, J.W.T. Chronic Inhibition of Na+/H+-Exchanger Attenuates Cardiac Hypertrophy and Prevents Cellular Remodeling in Heart Failure. Cardiovasc. Res. 2005, 65, 83–92.
- Inhibiting Mitochondrial Na+/Ca2+ Exchange Prevents Sudden Death in a Guinea Pig Model of Heart Failure|Circulation Research. Available online: https://www.ahajournals.org/doi/10.1161/CIRCRESAHA.115.303062 (accessed on 24 September 2023).
- Baartscheer, A.; Schumacher, C.A.; Wüst, R.C.I.; Fiolet, J.W.T.; Stienen, G.J.M.; Coronel, R.; Zuurbier, C.J. Empagliflozin Decreases Myocardial Cytoplasmic Na+ through Inhibition of the Cardiac Na+/H+ Exchanger in Rats and Rabbits. Diabetologia 2017, 60, 568–573.
- Uthman, L.; Baartscheer, A.; Bleijlevens, B.; Schumacher, C.A.; Fiolet, J.W.T.; Koeman, A.; Jancev, M.; Hollmann, M.W.; Weber, N.C.; Coronel, R.; et al. Class Effects of SGLT2 Inhibitors in Mouse Cardiomyocytes and Hearts: Inhibition of Na+/H+ Exchanger, Lowering of Cytosolic Na+ and Vasodilation. Diabetologia 2018, 61, 722–726.
- Li, X.; Lu, Q.; Qiu, Y.; do Carmo, J.M.; Wang, Z.; da Silva, A.A.; Mouton, A.; Omoto, A.C.M.; Hall, M.E.; Li, J.; et al. Direct Cardiac Actions of the Sodium Glucose Co-Transporter 2 Inhibitor Empagliflozin Improve Myocardial Oxidative Phosphorylation and Attenuate Pressure-Overload Heart Failure. J. Am. Heart Assoc. 2021, 10, e018298.
- SGLT2 Inhibitors and the Cardiac Na+/H+ Exchanger-1: The Plot Thickens | Cardiovascular Research|Oxford Academic. Available online: https://academic.oup.com/cardiovascres/article/117/14/2702/6288488?login=false (accessed on 24 September 2023).
- Baker, H.E.; Kiel, A.M.; Luebbe, S.T.; Simon, B.R.; Earl, C.C.; Regmi, A.; Roell, W.C.; Mather, K.J.; Tune, J.D.; Goodwill, A.G. Inhibition of Sodium–Glucose Cotransporter-2 Preserves Cardiac Function during Regional Myocardial Ischemia Independent of Alterations in Myocardial Substrate Utilization. Basic. Res. Cardiol. 2019, 114, 25.
- Chang, C.; Su, H.; Zhang, D.; Wang, Y.; Shen, Q.; Liu, B.; Huang, R.; Zhou, T.; Peng, C.; Wong, C.C.L.; et al. AMPK-Dependent Phosphorylation of GAPDH Triggers Sirt1 Activation and Is Necessary for Autophagy upon Glucose Starvation. Mol. Cell 2015, 60, 930–940.
- Ghosh, H.S.; McBurney, M.; Robbins, P.D. SIRT1 Negatively Regulates the Mammalian Target of Rapamycin. PLoS ONE 2010, 5, e9199.