Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biology
Hutchinson–Gilford progeria syndrome (HGPS) is an extremely rare genetic disorder caused by the mutant protein progerin, which is expressed by the abnormal splicing of the LMNA gene. HGPS affects systemic levels, with the exception of cognition or brain development, in children, showing that cellular aging can occur in the short term.
- Hutchinson–Gilford progeria syndrome
- progerin
1. Introduction
Hutchinson–Gilford progeria syndrome (HGPS; OMIM#176670) was first reported more than 100 years ago by Hutchinson and Gilford in 1886 and 1897, respectively [1]. The condition was designated as a premature aging syndrome by Gilford based on the fact that the symptoms associated with aging are similar to the changes seen in older people in general, including a lack of subcutaneous fat, hair loss, joint contractures, a progressive cardiovascular disease similar to atherosclerosis, and death from heart attacks and strokes in childhood. As patients typically live to their teens or early 20s and usually die before reaching reproductive age, this syndrome is not inherited. Diagnosis can be achieved within the first 6 months of age, although prominent and noticeable symptoms may be observed later [2][3][4][5][6][7][8]. This fatal pediatric disease remained a medical mystery until genetic mapping revealed that 90% of patients have a de novo point mutation in the LMNA gene that replaces cytosine with thymine [9][10].
Nuclear membrane proteins (lamins A and C), encoded by the LMNA gene, are structural components of the nuclear lamina, a network of proteins underneath the nuclear membrane that determines the shape and size of the nucleus [11][12]. LMNA produces four proteins as a result of alternative splicing: lamin A and lamin C as major products, and lamin C2 and lamin A delta 10 as minor products. Lamins A and C are similar through the first 566 amino acids (encoded by exons 1–10) but deviate at the carboxyl terminus [3][13][14]. Prelamin A, but not lamin C, contains a CaaX motif at its C-terminus and undergoes farnesylation and methylation (Figure 1). Lamin A is synthesized as a precursor (prelamin A) and matures through four sequential post-translational processing steps [15]. First, farnesyltransferase (FTase) adds a 15-carbon farnesyl moiety to the carboxyl-terminal cysteine. Second, the Zmpste24 endopeptidase cleaves the last three amino acids of prelamin A. Third, the newly exposed farnesyl cysteine is carboxyl-methylated by a prenyl protein-specific methyltransferase. Finally, the endopeptidase removes 15 carboxyl-terminal amino acids from the protein, resulting in the release of mature lamin A.
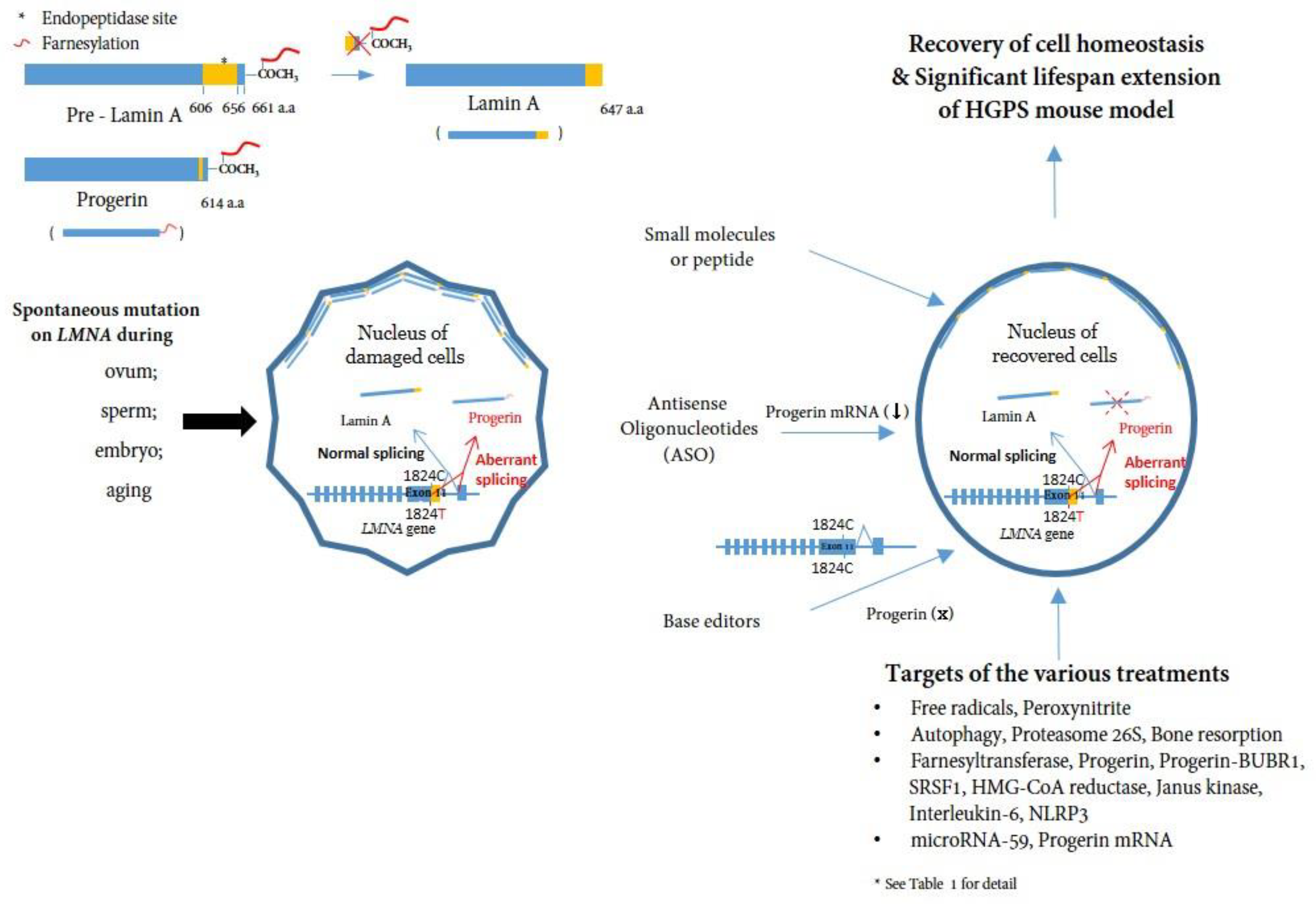
Figure 1. A schematic representation of the post-translational processing of lamin A and progerin and the effect of progerin inhibition on cells. Spontaneous mutations in LMNA (c. 1824C>T) in eggs, sperm, embryos, or during aging cause alternative splicing of the LMNA gene, leading to the accumulation of progerin in the nuclear layer. Consequently, the accumulation of progerin renders the cell unhealthy in a mechanophysiological manner. Inhibiting progerin by several methods (small molecules, ASOs, base editors) can restore damaged cells (See Table 1 for detail).
It has been questioned whether the post-translational processing steps of prelamin A are essential in targeting the protein to the nuclear envelope [16]. Mice that could directly produce mature lamin A without going through the usual prelamin A synthesis and processing steps were created. However, no detectable disease phenotype was observed in the mice and the nuclear membrane of mature lamin A appeared normal [16], suggesting that prelamin A processing is minimally important for the nuclear targeting of mature lamin A and is independent of lamin B in laboratory mice.
HGPS belongs to a group of diseases called laminopathies, in which mutations across the LMNA gene result in a wide range of overlapping disorders [17]. Genetic mapping of the genome from patients elucidated that a sporadic, autosomal-dominant de novo point mutation, c.1824C>T (p.G608G) (NM_170707.3) in exon 11 of the human LMNA gene, mediates abnormal alternative splicing [9][10], which produces an abnormal variant protein called progerin, which is responsible for this accelerated aging disease [18][19][20] (Figure 1).
2. Absence of Primary Neurological Disease in Hutchinson–Gilford Progeria Syndrome
Extensive studies have been conducted to examine mutations in the LMNA gene encoding prelamin A and lamin C, which result in distinct muscular dystrophy, cardiomyopathy, partial lipodystrophy, and progeroid syndromes. These laminopathies mostly affect mesenchymal tissues (e.g., the myocardium, skeletal muscle, adipose tissue, fibrous connective tissue, and bone tissues). However, one confusing observation in patients with HGPS is that they generally show fundamental and dramatic premature aging but do not exhibit any noticeable cognitive damage. For many years, it has been puzzling that patients with HGPS do not have any primary neurological disease. However, recent research has confirmed a lack of lamin A expression, the major isoform of LMNA, in HGPS patient-driven induced pluripotent stem cells (iPSCs) [21][22][23][24]. In most tissues, the amounts of lamins A and C are approximately equal; however, the brain mostly generates lamin C and very little lamin A [21][25]. Immunohistochemistry has indicated that lamin C is expressed at high levels in the neurons and glia of the brain, but the expression of prelamin A and lamin A is restricted to the vascular endothelial cells [25]. Further studies have shown that the expression of prelamin A in the brain is downregulated by miR-9, a microRNA highly expressed in the brain that binds to a single site in the 3′ untranslated region of prelamin A [21][22][25][26]. The ectopic expression of miR-9 in fibroblasts or HeLa cells decreases the levels of prelamin A and lamin A proteins but does not influence lamin C expression [25]. In lamin-A-only knock-in mice, where there is no alternative splicing and the output of all genes is directed to the prelamin A transcript, high levels of lamin A are found in the peripheral tissues, but very little lamin A is found in the brain [25]. Likewise, a knock-in mouse was created to direct the production of LMNA towards the progerin transcript. In this model, high levels of progerin were expressed in peripheral tissues, while minimal levels were observed in the brain [25], demonstrating that the unique expression pattern of lamin A/lamin C in the brain is not the result of alternative splicing. This regulation of lamin A in the brain provides us with a basis to further study improperly processed progerin and toxic lamin A. Children with HGPS have aging-like phenotypes in many tissues but lack common features of physiological aging in the central nervous system (CNS), such as senile dementia. Therefore, progerin accumulation in cells is considered a pathology-inducing factor.
Additionally, several studies have highlighted the important role of nuclear lamins in the CNS, indicating that type B lamins, lamins B1/B2, play an important role in neuronal migration in the developing brain [27][28][29]. Duplication of the LMNB1 gene encoding lamin B1 has been shown to cause autosomal-dominant leukodystrophy (ADLD) [30][31]. More recently, both LMNB1 deficiency and overexpression have been reported to inhibit proliferation, but only LMNB1 overexpression induces senescence, which is prevented by telomerase expression or p53 inactivation. A concomitant decrease in lamin A/C levels aggravates this phenotype. These findings show that changes in the expression of LMNB1 inhibit proliferation and are potentially relevant in understanding the molecular pathophysiology of ADLD [32], suggesting the possibility that a distinct spectrum of “brain laminopathies” might eventually be mapped to missense mutations in LMNB, not in LMNA.
This entry is adapted from the peer-reviewed paper 10.3390/cells12182299
References
- Gilford, H. Progeria: A form of senilism. Practitioner 1904, 73, 188–217.
- Merideth, M.A.; Gordon, L.B.; Clauss, S.; Sachdev, V.; Smith, A.C.; Perry, M.B.; Brewer, C.C.; Zalewski, C.; Kim, H.J.; Solomon, B.; et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N. Engl. J. Med. 2008, 358, 592–604.
- Burke, B.; Stewart, C.L. Life at the edge: The nuclear envelope and human disease. Nat. Rev. Mol. Cell Biol. 2002, 3, 575–585.
- Kipling, D.; Davis, T.; Ostler, E.L.; Faragher, R.G. What can progeroid syndromes tell us about human aging? Science 2004, 305, 1426–1431.
- Miller, R.A. “Accelerated aging”: A primrose path to insight? Aging Cell 2004, 3, 47–51.
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217.
- Gordon, L.B.; Rothman, F.G.; López-Otín, C.; Misteli, T. Progeria: A paradigm for translational medicine. Cell 2014, 156, 400–407.
- Ahmed, M.S.; Ikram, S.; Bibi, N.; Mir, A. Hutchinson-Gilford progeria syndrome: A premature aging disease. Mol. Neurobiol. 2018, 55, 4417–4427.
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin a cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298.
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, C.L.; Munnich, A.; Le Merrer, M.; et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003, 300, 2055.
- Lin, F.; Worman, H.J. Structural organization of the human gene encoding nuclear lamin a and nuclear lamin c. J. Biol. Chem. 1993, 268, 16321–16326.
- Wydner, K.L.; McNeil, J.A.; Lin, F.; Worman, H.J.; Lawrence, J.B. Chromosomal assignment of human nuclear envelope protein genes LMNA, LMNB1, and LBR by fluorescence in situ hybridization. Genomics 1996, 32, 474–478.
- Fisher, D.Z.; Chaudhary, N.; Blobel, G. CDNA sequencing of nuclear lamins a and c reveals primary and secondary structural homology to intermediate filament proteins. Proc. Natl. Acad. Sci. USA 1986, 83, 6450–6454.
- Mounkes, L.C.; Burke, B.; Stewart, C.L. The a-type lamins: Nuclear structural proteins as a focus for muscular dystrophy and cardiovascular diseases. Trends Cardiovasc. Med. 2001, 11, 280–285.
- Davies, B.S.; Fong, L.G.; Yang, S.H.; Coffinier, C.; Young, S.G. The posttranslational processing of prelamin a and disease. Annu. Rev. Genom. Hum. Genet. 2009, 10, 153–174.
- Coffinier, C.; Jung, H.J.; Li, Z.; Nobumori, C.; Yun, U.J.; Farber, E.A.; Davies, B.S.; Weinstein, M.M.; Yang, S.H.; Lammerding, J.; et al. Direct synthesis of lamin a, bypassing prelamin a processing, causes misshapen nuclei in fibroblasts but no detectable pathology in mice. J. Biol. Chem. 2010, 285, 20818–20826.
- Worman, H.J.; Bonne, G. “Laminopathies”: A wide spectrum of human diseases. Exp. Cell Res. 2007, 313, 2121–2133.
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of mutant lamin a causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8963–8968.
- McClintock, D.; Gordon, L.B.; Djabali, K. Hutchinson-Gilford progeria mutant lamin a primarily targets human vascular cells as detected by an anti-lamin a g608g antibody. Proc. Natl. Acad. Sci. USA 2006, 103, 2154–2159.
- Kashyap, S.; Shanker, V.; Sharma, N. Hutchinson-Gilford progeria syndrome: A rare case report. Indian Dermatol. Online J. 2014, 5, 478–481.
- Nissan, X.; Blondel, S.; Navarro, C.; Maury, Y.; Denis, C.; Girard, M.; Martinat, C.; De Sandre-Giovannoli, A.; Levy, N.; Peschanski, M. Unique preservation of neural cells in Hutchinson- Gilford progeria syndrome is due to the expression of the neural-specific mir-9 microRNA. Cell Rep. 2012, 2, 1–9.
- Young, S.G.; Jung, H.J.; Lee, J.M.; Fong, L.G. Nuclear lamins and neurobiology. Mol. Cell Biol. 2014, 34, 2776–2785.
- Liu, G.H.; Barkho, B.Z.; Ruiz, S.; Diep, D.; Qu, J.; Yang, S.L.; Panopoulos, A.D.; Suzuki, K.; Kurian, L.; Walsh, C.; et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature 2011, 472, 221–225.
- Zhang, J.; Lian, Q.; Zhu, G.; Zhou, F.; Sui, L.; Tan, C.; Mutalif, R.A.; Navasankari, R.; Zhang, Y.; Tse, H.F.; et al. A human iPSC model of Hutchinson Gilford progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 2011, 8, 31–45.
- Jung, H.J.; Coffinier, C.; Choe, Y.; Beigneux, A.P.; Davies, B.S.; Yang, S.H.; Barnes, R.H., 2nd; Hong, J.; Sun, T.; Pleasure, S.J.; et al. Regulation of prelamin a but not lamin c by mir-9, a brain-specific microRNA. Proc. Natl. Acad. Sci. USA 2012, 109, E423–E431.
- Jung, H.J.; Tu, Y.; Yang, S.H.; Tatar, A.; Nobumori, C.; Wu, D.; Young, S.G.; Fong, L.G. New LMNA knock-in mice provide a molecular mechanism for the ‘segmental aging’ in Hutchinson-Gilford progeria syndrome. Hum. Mol. Genet. 2014, 23, 1506–1515.
- Coffinier, C.; Chang, S.Y.; Nobumori, C.; Tu, Y.; Farber, E.A.; Toth, J.I.; Fong, L.G.; Young, S.G. Abnormal development of the cerebral cortex and cerebellum in the setting of lamin b2 deficiency. Proc. Natl. Acad. Sci. USA 2010, 107, 5076–5081.
- Coffinier, C.; Fong, L.G.; Young, S.G. Lincing lamin b2 to neuronal migration: Growing evidence for cell-specific roles of b-type lamins. Nucleus 2010, 1, 407–411.
- Coffinier, C.; Jung, H.J.; Nobumori, C.; Chang, S.; Tu, Y.; Barnes, R.H., 2nd; Yoshinaga, Y.; de Jong, P.J.; Vergnes, L.; Reue, K.; et al. Deficiencies in lamin b1 and lamin b2 cause neurodevelopmental defects and distinct nuclear shape abnormalities in neurons. Mol. Biol. Cell 2011, 22, 4683–4693.
- Padiath, Q.S.; Fu, Y.H. Autosomal dominant leukodystrophy caused by lamin b1 duplications a clinical and molecular case study of altered nuclear function and disease. Methods Cell Biol. 2010, 98, 337–357.
- Padiath, Q.S.; Saigoh, K.; Schiffmann, R.; Asahara, H.; Yamada, T.; Koeppen, A.; Hogan, K.; Ptácek, L.J.; Fu, Y.H. Lamin b1 duplications cause autosomal dominant leukodystrophy. Nat. Genet. 2006, 38, 1114–1123.
- Dreesen, O.; Chojnowski, A.; Ong, P.F.; Zhao, T.Y.; Common, J.E.; Lunny, D.; Lane, E.B.; Lee, S.J.; Vardy, L.A.; Stewart, C.L.; et al. Lamin b1 fluctuations have differential effects on cellular proliferation and senescence. J. Cell Biol. 2013, 200, 605–617.
This entry is offline, you can click here to edit this entry!