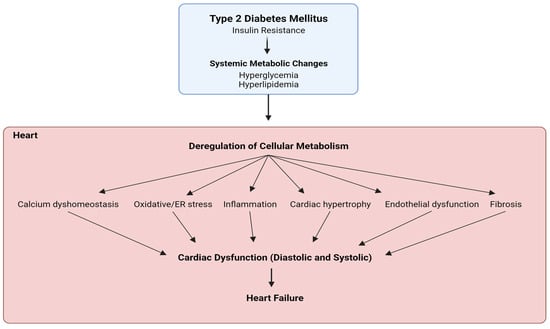
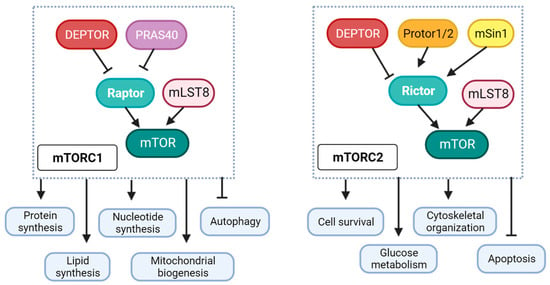
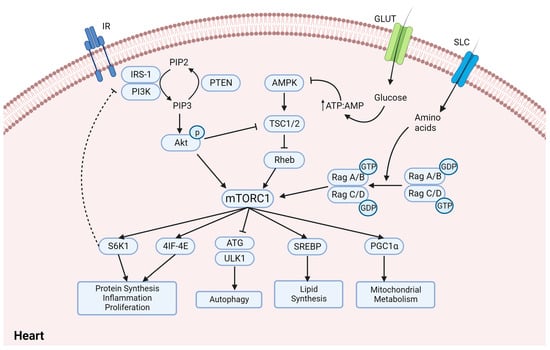
Ionic imbalance, including a state of calcium overload as well as increased intracellular sodium, is a key player in the development of cardiac dysfunction and characteristics of diabetic cardiomyopathy [
58,
59,
60]. mTOR signaling is involved in diverse biological pathways by regulating ionic homeostasis, specifically by regulating the activity and expression of various Ca
2+ channels [
61,
62,
63]. Intertwined links between sarcoplasmic reticulum calcium homeostasis and mTORC1 signaling are critical for physiological and pathological cardiac hypertrophy [
64,
65]. Inhibition of mTORC1 with rapamycin induces Ca
2+ release from lysosomes through the activation of two-pore segment channel 2, TPC2 [
66]. The downregulation of the inward rectifier potassium (I
K1) channel with intracellular Ca
2+ overload is a hallmark in cardiac hypertrophy, interstitial fibrosis, and electrical remodeling and failure [
67]. Specifically, mTORC1 regulates the lysoNa
ATP, which determines the sensitivity of endolysosome’s resting membrane potential to Na
+ and cytosolic ATP as well as controls lysosomal pH stability and whole-body amino acid homeostasis [
68,
69]. Selective I
K1 agonist attenuates cardiac remodeling by promoting autophagy via negatively regulating calcium-activated CaMKII and mTOR signaling [
69]. The activation of I
K1 channel protects the heart against myocardial ischemia-induced cardiac dysfunction by inhibiting mTOR-p70S6 signaling pathway [
68]. mTOR also acutely controls endosomal and lysosomal functions through the endolysosomal ATP-sensitive Na
+ channel (lysoNa
ATP) in response to changes under different nutrition status and metabolic conditions [
62]. mTORC1 regulates the endolysosomal ATP-sensitive Na
+ channel (lysoNa
ATP), which determines the sensitivity of endolysosome’s resting membrane potential to Na
+ and cytosolic ATP as well as controls lysosomal pH stability and whole-body amino acid homeostasis.
3. Sodium and Glucose Co-Transporter Inhibitors (SGLT2is)—Do They Regulate mTORC1 in Diabetic Cardiomyopathy?
Despite the boom in preclinical and clinical studies on diabetic cardiomyopathy, its pathogenesis and unified paradigm remain unclear for devising specific therapeutic strategies. As a result, a plethora of research is still based on figuring out the most effective therapeutic approach to treating diabetes, diabetic cardiomyopathy, and related heart failure. To fill this critical gap in therapeutics, mTORC1 might be a key target for diabetic therapeutics, which also addresses other co-morbidities like cardiomyopathy in a glycemic and non-glycemic context. In T2D, metformin is usually the first-line standard treatment [
87] and though a few studies have shown metformin to exert cardioprotective effects via mTORC1 regulation [
57], these reports are very limited despite a long history of metformin use [
88].
Glucose homeostasis, a crucial diabetes parameter associated with mTORC1 [
93], is actively regulated by transporters like GLUTs and SGLTs that mediate D-glucose transport. While SLC2 genes encode for facilitated glucose diffusion transporters GLUT, sodium glucose co-transporters SGLT1-5 are encoded by SLC5. SGLT transports glucose into cells via Na
+/K
+-ATPase pump gradient and two major SGLT isoforms are SGLT1 and SGLT2. SGLT1 is expressed in the small intestine, kidneys, brain, and heart, while SGLT2 is expressed in kidney and pancreatic β cells. SGLT1 primarily acts as rate limiting intestinal glucose absorption whereas SGLT2 manages bulk glucose reabsorption in the kidneys [
94]. In the kidneys, SGLT2 in the S1/S2 segment of the convoluted proximal tubule in kidney nephrons reabsorbs 90% of the glomerular filtrate glucose, aided by a positive sodium gradient [
95], while SGLT1 reabsorbs the remaining 10% in the S3 segment of the proximal tubule [
96]. The reabsorbed glucose in the tubular epithelial cells is flushed back into circulation through GLUT2 and this whole unidirectional transport of glucose is coupled to and regulated by the Na
+K
+ ATPase pump on the basolateral side of the cells [
97]. SGLT2is primarily work as anti-hyperglycemic agents by blocking the SGLT2 channel and preventing glucose reabsorption, thus preventing hyperglycemia in diabetes mellitus.
Over the years, besides significant glucose-lowering efficacy, SGLT2is have also shown remarkable cardiovascular benefits in renal-impaired patients with lower glomerular filtration rates, indicating a major role of SGLT2is in promoting diabetic cardiac dysfunction benefits [
101].
Although SGLT2is have shown interesting benefits in terms of cardiac function with or without T2D, the expression of SGLT2 channels is negligible in the heart, further highlighting SGLT2-independent functions of these inhibitors in diabetic cardiomyopathy and cardiac dysfunction [
134,
135]. The presence of SGLT1 in the heart is well established and is reportedly overexpressed in T2DM patients [
136]. Along with glucose transporters (GLUT), SGLT1 is involved in cardiomyocyte glucose uptake, and in ischemic conditions, SGLT1 is reported to supplement the ATP reserve by increasing glucose utilization, thus playing a role in myocardial energy metabolism [
137,
138].
Amidst some theories to explain the role of SGLT2is in cardiac function that involves the renin–angiotensin–aldosterone system (RAAS) and diuretic pathways [
149], a prominent hypothesis to support the role of SGLT2is in the heart concerns the involvement of sodium ion homeostasis [
150]. Cardiac Na
+ and Ca
2+ homeostasis plays a major role in maintaining heart physiology, rhythm, and contraction [
151]. Increased intracellular sodium (Na
i+) due to hyperactive sodium hydrogen exchanger (NHE) is well known in cardiac dysfunction pathologies and leads to elevated Ca
2+ efflux from the mitochondria, resulting in oxidative stress and deteriorated cellular function [
152,
153]. Besides variable degrees of cross-reactivity with SGLT1 [
154], several reports on SGLT2is have demonstrated non-SGLT2 cardiac-based off-target effects in reducing ventricular myocyte systolic Ca
2+ and lowering myocardial cytoplasmic Na
+/Ca
2+ via NHE regulation [
155]. A reduction in myocardial Na
i+ via the inhibition of Na
+/H
+ or Na
+/Ca
2+ exchangers has been reported to improve cardiac hypertrophy and heart failure [
156,
157]. Preclinical studies have shown that SGLT2is can directly bind and reduce NHE activity in cardiomyocytes, leading to decreased Na
i+ and restored Ca
2+ homeostasis, resulting in improved cardiac function and antioxidative capacity of cardiomyocytes [
158,
159], but the observations are not consistent [
148,
160].
From another perspective, SGLT2 inhibitors have been shown in some studies to promote ketogenesis, lipid oxidation, and erythrocytosis, which can reflect a fasting-like transcriptional paradigm by mimicking nutrient deprivation and hypoxia [
166] but can also be responsible for the moderate adverse effects in SGLT2i clinical trials. Although systemic glucose lowering or ketogenesis by SGLT2is in vivo can modulate a starvation and nutrient deprivation environment, a possible GLUT inhibition by selective SGLT2is might be responsible for glucose deprivation in isolated cardiomyocytes. A depletion in the glucose environment caused by SGLT2is can reduce the ATP/ADP ratio, leading to increased AMP that stimulates the phosphorylation of AMPK and subsequently, phosphorylates GAPDH to activate SIRT1 [
167]. AMPK (nutrient sensor) and SIRT1 (redox rheostat) activation help cardiomyocytes to adapt in response to SGLT2i-mediated nutrient-deprived conditions. AMPK and SIRT1 activation have been reported to negatively regulate mTORC1 in a Tsc1/2-dependent manner [
49,
168], thus indicating that SGLT2is indirectly inactivate mTORC1 by mimicking a nutrition-deprivation setting. Furthermore, a recent study of SGLT2is in obesity-related diabetic cardiomyopathy reported sestrin2-mediated AMPK activation/mTORC1 inactivation in cardiomyocytes upon empagliflozin treatment [169]. Some studies have reported energy deprivation as a cause for sestrin2 activation [170], which can be correlated with AMPK/mTORC2 activation besides inhibiting mTORC1 and promoting autophagy [171]. This adds another revelation to the role of SGLT2is in attenuating diabetic cardiomyopathy conditions by promoting an energy-deprived environment. A recent study by Zhang et al. in 2023 suggested that empagliflozin significantly reduced diabetic cardiomyopathy by promoting branched-chain amino acid catabolism, inhibiting mTORC1/p-ULK1, and reactivating autophagy [172], while another study by Feng et al. reported that dapagliflozin prevented cardiac dysfunction in diabetic rats by restoring autophagy by repressing mTORC1 and activating AMPK [173].
Figure 4 shows a summary of SGLT2 inhibitor mechanisms to target renal glucose absorption (canonical) and cardiac mTORC1 signaling (non-canonical) in diabetic cardiomyopathy.
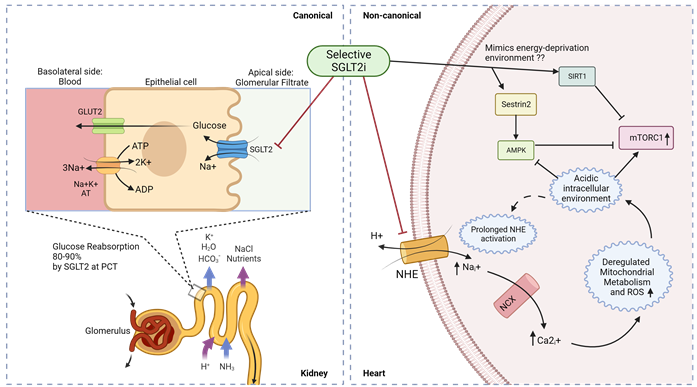
Figure 4. Canonical and non-canonical (cardiac) mechanism for SGLT2i function in diabetic cardiomyopathy. Luminal glucose in the renal glomerular filtrate is reabsorbed (80–90%) into the proximal tubular epithelial cells via SGLT2 in the S1 segment of proximal convoluted tubule (PCT) of nephrons, and subsequently transported to the basolateral interstitial fluid through GLUT2. In diabetic cardiomyopathy therapy, SGLT2is act in a canonical manner by preventing renal glucose reabsorption to reduce overall hyperglycemia but studies indicate a non-canonical mechanism of SGLT2is in the heart. Owing to the absence of SGLT2 channels in the heart, selective SGLT2is can act on prolonged activated NHE to reduce intracellular sodium ions and subsequently promote an alkaline intracellular pH, which has been reported to activate AMPK and inhibit mTORC1. SGLT2 inhibitors have also been reported to mimic an energy-deprived environment that might promote Sestrin2-mediated AMPK and SIRT1 activation, leading to mTORC1 inactivation and cardiovascular benefits. PCT—proximal convoluted tubule; ; NHE—sodium–hydrogen exchanger; NCX—sodium–calcium exchanger.
4. Conclusions
Cardiovascular diseases like diabetic cardiomyopathy are critical co-manifestations in patients with diabetes mellitus and several medical approaches are currently targeting diabetes with a significant consideration for treatments that also improve cardiac dysfunction and cardiomyopathy. A thorough analysis of T2D and its co-morbidities, in both glycemic and non-glycemic contexts, translates into mTORC1 being a unified therapeutic target. Besides the standard metformin treatment for diabetes, SGLT2 inhibitors have come up recently as potential anti-diabetic drugs that show very promising cardiovascular protection. Although the mechanisms of the cardiac effects of SGLT2is are still being explored, the existing hypotheses consistently point to mTORC1 regulation via ionic dyshomeostasis/stress and mimics of nutrient deprivation. While the role of SGLT2is in downregulating mTORC1 is very critical for cardioprotective effects in diabetic patients, owing to the evidence of improved cardiac function by mTORC1-inhibitor rapamycin, all roads do not lead to Rome; several studies have indicated that a fine balance between mTORC2 activation and mTORC1 inhibition is optimal for cardiac benefits in patients with/without diabetes. An in-depth understanding of off-target SGLT2i effects might open up novel treatment regimes with SGLT2is in several other cardiac, renal, pancreatic, cerebral, and hepatic pathologies as a single drug or combination therapy. With further clinical progress in exploring the mechanisms of how SGLT2is regulate cell signaling, drug modifications might also help in bypassing the adverse effects of genital infections, thus ameliorating the standard of patient care. Therefore, further research is pivotal to understand novel mechanisms of SGLT2is in the non-glycemic SGLT2-independent context besides shedding light on the role of SGLT2is in regulating the mTORC1/mTORC2 switch for a holistic approach towards diabetic cardiomyopathy and related metabolic disorders.