Therapy for inflammatory and autoimmune eye diseases focuses on managing the underlying condition and reducing inflammation in the eyes [
2]. Treatment plans vary depending on the specific condition and severity of the disease [
2]. Eye drops or ointments containing corticosteroids, non-steroidal anti-inflammatory drugs (NSAIDs), or immunosuppressive agents are usually applied directly to the eye to reduce mild inflammation and alleviate conjunctivitis and keratitis-related symptoms [
3]. In more severe cases, oral medications such as corticosteroids, immunosuppressants, or biologics may be used to suppress detrimental immune responses and to prevent the progression of ongoing eye inflammation [
3]. For certain conditions, such as severe uveitis, injections of corticosteroids or immunosuppressive agents may be administered directly into the eye. In some cases, surgical procedures such as vitrectomy or cataract surgery may be necessary to restore vision and to treat complications caused by inflammatory or autoimmune eye diseases [
3]. Although therapeutic strategies for the treatment of inflammatory eye diseases have evolved significantly, there are still some issues that limit the therapeutic efficacy of these approaches [
2,
3]. Prolonged use of corticosteroids may increase the risk of cataracts or glaucoma while long-term use of immunosuppressive drugs may inhibit the immune system throughout the body, increasing the risk of infections. Since inflammatory and autoimmune eye diseases manifest differently in each individual, identifying the most suitable treatment approach often requires a personalized approach [
2,
3]. Finally, achieving complete remission or long-term control of the underlying autoimmune or inflammatory disease can be challenging. Some patients may require ongoing treatment and monitoring to manage their symptoms and prevent relapses. Accordingly, a large number of experimental and clinical studies are continuously conducted in order to resolve limits and to improve therapeutic options for the treatment of inflammatory eye diseases [
2,
3].
2. Activation of NLRP3 Inflammasome in Eye-Infiltrated Immune Cells as an Initial Step in the Development of Inflammatory Eye Disease
The activation of the NLRP3 inflammasome in eye-infiltrated immune cells leads to the enhanced production of pro-inflammatory cytokines (interleukin-1β (IL-1β) and IL-18), which elicit inflammatory processes in ocular structures [
10].
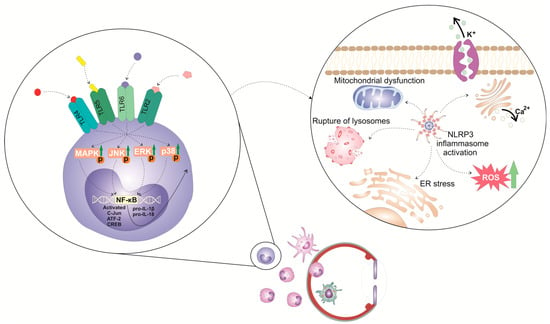
The activation of the NLRP3 inflammasome in immune cells is a tightly regulated, multi-step process (
Figure 1). It requires two steps: priming and activation. Since some immune cells do not naturally express pro-IL-1 and have inadequate amounts of NLRP3 for inflammasome activation, one of the main functions of the priming step is to stimulate the transcriptional production of NLRP3, pro-IL-1β, and pro-IL-18. This initial step involves the activation of transcriptional factors (the nuclear factor kappa B (NF-κB), c-Jun, ATF-2, and CREB) that, upon activation, translocate to the nucleus to induce the enhanced transcription of pro-inflammatory genes, resulting in the increased synthesis of NLRP3 and pro-IL-1β. The priming step of NLRP3 inflammasome activation is elicited by the activation of pattern recognition receptors (PRRs), including Toll-like receptors (TLR). These membrane-bound receptors are expressed on immune cells where recognized pathogen-associated molecular patterns (PAMPs) of microbial antigens or damage-associated molecular patterns (DAMPs) are released from injured cells [
10,
11]. Signals generated from activated PRRs, particularly TLR-2, TLR-4, TLR-5, and TLR-6, initiate phosphorylation and the consequent activation of extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 mitogen-activated protein kinase (MAPK), which, in turn, phosphorylate c-Jun, ATF-2, and CREB transcription factors that bind to the promoter regions of NLRP3 and pro-IL-1β, leading to their transcriptional upregulation [
10,
11]. Additionally, upon binding to TLRs, TLR ligands elicit the MyD88-driven activation of IL-1R-associated kinase 1 (IRAK-1), which, in a TNFR-associated factor 6 (TRAF6)-dependent manner, induces the activation of transforming growth factor-beta-activated kinase 1 (TAK-1), a key protein kinase responsible for the optimal activation of NF-κB. Activated TAK-1 phosphorylates and activates the IκB kinase (IKK) complex, which consists of IKKα, IKKβ, and IKKγ (also known as NEMO). The activated IKK complex phosphorylates the Inhibitor of κB (IκB), which, within cytosol, binds to NF-κB and prevents its nuclear translocation. The phosphorylation of IκB leads to its degradation via the ubiquitin–proteasome pathway. This degradation releases NF-κB, allowing it to translocate into the nucleus. Once in the nucleus, NF-κB binds to specific DNA sequences called κB sites, promoting the transcription of NF-κB-dependent genes, such as NLRP3, pro-IL-1β, and pro-IL-18, which are necessary for inflammasome activation.
Figure 1. Molecular mechanisms and signaling pathways involved in the activation of NLRP3 inflammasome in eye-infiltrated immune cells. The activation of the NLRP3 inflammasome in immune cells is a tightly regulated, multi-step process. It requires 2 steps: priming and activation. The priming step of NLRP3 inflammasome activation is elicited by the activation of pattern recognition receptors (PRRs), including Toll-like receptors (TLR). These membrane-bound receptors are expressed on immune cells that recognize pathogen-associated molecular patterns (PAMPs) of microbial antigens or damage-associated molecular patterns (DAMPs) released from injured cells. Signals generated from activated TLR-2, TLR-4, TLR-5, and TLR-6 initiate phosphorylation and consequent activation of extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 mitogen-activated protein kinase (MAPK), which, in turn, phosphorylate c-Jun, ATF-2, and CREB transcription factors that bind to the promoter regions of NLRP3 and pro-IL-1β, leading to their transcriptional upregulation.
In addition to the priming step, a second step, often referred to as the “activation signal” or the “danger signal” is necessary for the full and optimal activation of the NLRP3 inflammasome. This second step is driven by NLRP3 agonists, which activate NLRP3 to cause inflammasome assembly and mature IL-1 production. It can be triggered by a variety of stimuli, including microbial components, extracellular matrix breakdown products, and environmental antigens [
10,
11,
12]. The exact mechanisms by which these stimuli activate the NLRP3 inflammasome are still not fully understood, but proposed mechanisms include alterations in potassium (K
+) efflux, lysosomal rupture, mitochondrial dysfunction, and enhanced reactive oxygen species (ROS) production [
10,
11]. Increased intracellular levels of ATP, massive accumulation of uric acid crystals, microbial antigens, and environmental irritants can induce potassium (K
+) efflux from the immune cell, leading to decreased intracellular potassium levels. NLRP3 contains a potassium (K
+)-sensing domain that undergoes conformational changes in response to altered potassium levels [
10,
11]. Accordingly, a decrease in intracellular potassium levels leads to conformational changes in the NLRP3 protein, allowing its oligomerization and consequent association with adaptor ASC protein made via homotypic interactions between the pyrin domains of these two molecules. NLRP3:ASC interaction leads to the formation of a large protein complex called the inflammasome [
10,
11]. NIMA-related kinase 7 (NEK7), an important component of the NLRP3 inflammasome complex, stabilizes the NLRP3 protein and promotes its interaction with ASC protein. Once the NLRP3 inflammasome complex is formed, the CARD domain of ASC protein interacts with the CARD domain of pro-caspase-1, facilitating its recruitment to the inflammasome complex [
12]. Afterward, pro-caspase-1 undergoes self-cleavage and activation, resulting in the formation of active caspase-1. Active caspase-1 then cleaves pro-IL-1β and pro-IL-18 into their mature forms, IL-1β and IL-18. These two cytokines are important inflammatory mediators that crucially contribute to the generation and propagation of all inflammatory eye diseases [
10,
11].
In addition to cytokine release, caspase-1 activation also leads to the cleavage of gasdermin D (GSDMD), an effector protein involved in the execution of pyroptosis, a highly inflammatory form of programmed cell death, which has an important pathogenic role in the development and progression of ocular inflammation [
13]. NLRP3 and caspase-1-dependent cleavage of GSDMD results in the formation of two distinct fragments of GSDMD protein: the N-terminal (GSDMD-N) and the C-terminal domains (GSDMD-C). Once released from the C-terminal domain, GSDMD-N oligomerizes and inserts into the lipid bilayer of the plasma membrane, leading to the formation of large pores called gasdermin pores [
13]. Gasdermin pores disrupt the integrity of the plasma membrane, causing the efflux of intracellular ions, cytoplasmic proteins, and pro-inflammatory cytokines (IL-1β and IL-18) into the extracellular space. IL-1β and IL-18 propagate and aggravate inflammation initially elicited by the activation of NLRP3 inflammasome and importantly contribute to the creation of a “positive inflammatory loop” in inflamed eyes that finally results in enhanced activation and the increased accumulation of inflammatory immune cells in inflamed eyes [
13].
3. Molecular Mechanisms Responsible for NLRP3/IL-1β/IL-18-Dependent Generation of Detrimental Immune Response in Inflamed Eyes
NLRP3/IL-1β/IL-18-dependent recruitment of immune cells is a complex process involving the activation of endothelial cells (ECs), the release of chemotactic factors, the modulation of adhesion molecules, and changes in vascular dynamics (
Table 1) [
9]. NLRP3-generated IL-1β and IL-18 induce increased expression of adhesion molecules (intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), E and P-selectins) on the membrane of ECs [
9]. Additionally, the activation of NLRP3 inflammasome in immune cells enhances the affinity and avidity of integrin–ligand interactions and facilitates firm adhesion of immune cells to ECs. Precisely, the NLRP3/IL-1β/IL-18 axis upregulates the expression of integrins (lymphocyte function-associated antigen-1 (LFA-1) and very late antigen-4 (VLA-4)) on immune cells, which interact with their ligands on ECs, further promoting the adhesion and subsequent transmigration of immune cells into the inflamed tissues of affected eyes [
9].
Table 1. The role of inflammatory cytokines and chemokines in the development and progression of NLRP3 inflammasome-driven eye inflammation.
The NLRP3/IL-1β/IL-18 signaling pathway promotes vasodilation and induces increased vascular permeability in inflamed eyes [
14]. Vasodilation leads to the widening of blood vessels, allowing more immune cells to flow closer to the site of inflammation [
14]. Increased vascular permeability allows immune cells to extravasate from the bloodstream into the tissue spaces, facilitating their recruitment and accumulation in the inflamed eyes [
14].
Additionally, the activation of NLRP3 inflammasome induces massive production of inflammatory cytokines (tumor necrosis factor alpha (TNF-α), IL-6, and IL-8), chemokines (CCL2, CCL3, CCL5, CXCL8, and CXCL10), and prostaglandins in eye-infiltrated monocytes and lymphocytes [
10,
11]. These inflammatory mediators act synergistically with IL-1β and IL-18 to promote the recruitment of inflammatory immune cells from the bloodstream to the site of eye inflammation [
10,
11].
Also, the NLRP3/IL-1β/IL-18 pathway importantly contributes to tissue remodeling in inflamed eyes [
18]. This axis stimulates the production of matrix metalloproteinases (MMPs) in immune cells, ECs, and fibroblasts. MMP-2, MMP-3, and MMP-9 degrade the extracellular matrix components, including collagen and proteoglycans, leading to tissue damage and breakdown of structural integrity [
18]. The NLRP3-dependent increase in intraocular concentration of IL-1β and IL-18 disrupts the integrity of the blood-aqueous barrier and blood-retinal barrier, which normally prevents the free passage of cells and molecules between the blood and the eye [
18]. In this way, NLRP3/IL-1β/IL-18 signaling facilitates the migration of immune cells across the barriers. This leads to increased vascular permeability and the leakage of fluid, proteins, and cells into the ocular tissues, exacerbating the ongoing inflammation. Also, the NLRP3/IL-1β/IL-18-dependent disruption of blood-aqueous and blood-retinal barriers may result in complications such as cystoid macular edema, retinal detachment, and glaucoma [
18].
Finally, the NLRP3/IL-1β/IL-18 axis is essential for shaping the adaptive immune response and for promoting effective T and B cell-driven immune defense against eye-invading pathogens [
19]. Accordingly, the dysregulation of the NLRP3/IL-1β/IL-18 pathway can lead to an aberrant adaptive immune response and contribute to the development of autoimmune diseases and chronic inflammation in the eyes [
19].
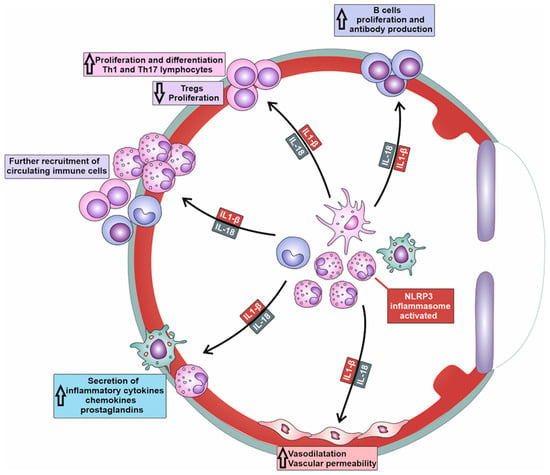
When the NLRP3 inflammasome is activated, IL-1beta and IL-18 are released, activating naive T cells and promoting their proliferation and clonal expansion (
Figure 2). The NLRP3/IL-1β/IL-18 axis can synergize with other co-stimulatory molecules, such as CD28, to amplify T cell activation signals [
10,
15]. NLRP3/IL-1β/IL-18 acts as a co-stimulatory signaling pathway that enhances the activation of T cell receptors (TCRs) on T cells, leading to increased production of cytokines and cytotoxic molecules. The NLRP3/IL-1β/IL-18-dependent activation of T cells contributes to the expansion of antigen-specific T cell responses and the generation of an effective adaptive immune response [
10,
11,
15].
Figure 2. Molecular mechanisms responsible for NLRP3-dependent generation of detrimental immune response in inflamed eyes. Activation of NLRP3 inflammasome results in increased production of IL-1β and IL-18 in tissue-infiltrated macrophages and neutrophils. IL-1β and IL-18 induce massive recruitment of circulating leukocytes in inflamed tissues, increased expansion of inflammatory Th1 and Th17 lymphocytes, enhanced production of inflammatory cytokines and chemokines in neutrophils and macrophages, attenuated proliferation of immunosuppressive T regulatory cells (Tregs), and increased antibody production in plasma cells.
Importantly, the NLRP3/IL-1β/IL-18 axis can influence the differentiation of T cells, particularly CD4+ T helper cells [
15]. IL-1β and IL-18 promote the differentiation of naïve CD4+ T cells in effector, pro-inflammatory T helper 1 (Th1) cells and Th17 cells, which are mainly responsible for the T cell-driven injury of corneas, conjunctiva, lacrimal, and meibomian glands in patients suffering from severe keratitis, uveitis, conjunctivitis, and DED [
15]. IL-1β and IL-18 act synergistically with other inflammatory (pro-Th1 and pro-Th17) cytokines such as IL-12 and IL-23 to induce the expression of lineage-specific transcription factors (e.g., T-bet for Th1 cells and RORγt for Th17 cells) and the production of effector cytokines (e.g., IFN-γ for Th1 cells and IL-17A for Th17 cells) [
10,
11,
15]. Th1 and Th17 lymphocytes are inflammatory cells that play a crucially important pathogenic role in the development of DED. Th1 cells produce IFN-γ, which increases the cytotoxic properties of NK cells and enhances the production of nitric oxide (NO), ROS, and inflammatory cytokines in macrophages and DCs, improving their antigen-presenting properties. Th17 cell-derived IL-17 generates inflammatory phenotypes in eye-infiltrated neutrophils and promotes neutrophil-driven tissue injury in inflamed eyes, lacrimal, and meibomian glands of DED patients [
10,
11,
15].
Additionally, the NLRP3/IL-1β/IL-18 axis may inhibit the generation and suppressive function of Tregs, which play a crucial role in immune tolerance within the eyes [
16]. NLRP3/IL-1β/IL-18 signaling can suppress the expression and activity of the transcription factor forkhead box P3, (Foxp3), which is critical for Treg development and function [
16]. By attenuating the generation of immunosuppressive Tregs, the NLRP3/IL-1β/IL-18 axis promotes the progression of detrimental immune response in inflamed eyes, crucially contributing to the aggravation of ongoing inflammation [
16].
In addition to cellular immunity, NLRP3/IL-1β/IL-18 signaling controls the activation of B cells and regulates antibody production, importantly contributing to the development of humoral immune response against microbial pathogens [
17]. The NLRP3/IL-1β/IL-18 axis initiates co-stimulatory signals for B cell activation, enhancing B cell receptor (BCR) downstream pathways and increasing the secretory profile of plasma cells [
17]. The NLRP3/IL-1β/IL-18 axis can induce the increased expression of activation-induced cytidine deaminase (AID), which is involved in class-switch recombination and somatic hypermutation, leading to the production of different antibody isotypes and high-affinity antibodies [
16,
17]. Accordingly, the NLRP3/IL-1β/IL-18 signaling pathway plays an important role in the development and progression of antibody-dependent autoimmune eye diseases, including Graves’ ophthalmopathy, Myasthenia gravis, Ocular Cicatricial Pemphigoid, and SLE-related DED [
15,
17].
In line with all these findings, the targeting of the NLRP3/IL-1β/IL-18 axis in immune cells has emerged as a potentially new therapeutic approach that could further enhance the efficacy of immunoregulatory agents, including MSCs, in the therapy of inflammatory and autoimmune eye disorders [
9].