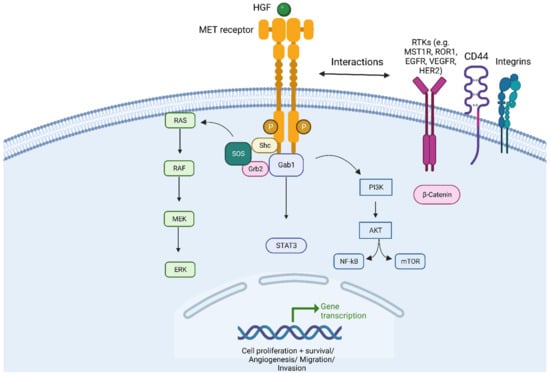
MET (mesenchymal-epithelial transition) is a proto-oncogene situated on chromosome 7q21-q31 that encodes for transmembrane receptor tyrosine kinase normally expressed on epithelial cells. The Hepatocyte Growth Factor (HGF)/c-Met pathway is well characterized and recognized for its essential role in several cell vital processes in embryonic development, in the repair of injured tissues, as well as in carcinogenesis and tumour progression [
1,
2,
3,
4,
5]. The HGF is produced by fibroblast cells, neutrophils and macrophages but was initially identified as a mitogen for hepatocytes [
6,
7]. Importantly, c-MET can interact with other ligands or cellular receptors, such as MET homolog RON (also known as MST1R), ROR1, CD44, integrins, and CD151, increasing the complexity of the pathway [
8] (
Figure 1). MET can be expressed in normal epithelial cells and overexpressed in several cancer cells, including Non-Small-Cell Lung Cancer (NSCLC) [
9,
10,
11]. MET can play its role in cancer progression in different circumstances when cancer cells harbour MET dysregulation (MET addiction) [
12,
13,
14]. MET alterations were involved in several cancers and consisted of MET mutations in renal papillary cancer (1–4%, first described in tumours), as well as MET amplification (MET AMP) in gastric cancer, melanoma (12%), de novo or as resistance to tyrosine kinase inhibitors in epidermal growth factor receptor (EGFR)-mutated NSCLC (15%), or colorectal cancer that develops resistance to EGFR antibodies [
12,
13,
15,
16,
17].
2. Prognostic Role of MET in NSCLC
Some retrospective studies of surgical series have reported the prognostic role of MET dysregulation in NSCLC, which is summarized in
Table 1 [
18,
19,
20,
21,
22,
23,
24,
25,
26,
27,
28,
29,
30].
Table 1. Summary of retrospectives surgical series investigating the prognostic role of MET dysregulation.
Reference: * Not reported, prognosis was assessed as 5-year overall survival. Abbreviations: Pt N = patient number; F-up = follow-up; ADK = adenocarcinoma; NSCLC = Non-Small-Cell Lung Cancer; ADS = adenosquamous carcinoma; IHC = immunohistochemistry; HGF = Hepatocyte Growth Factor; MET = mesenchymal–epithelial transition gene; b-MET mRNA = circulating MET mRNA; phospho-MET = MET phosphorylated; RT-PCR = real-time polymerase chain reaction; MET/HGF OE = MET/HGF overexpressed; GCN = gene copy number; WB = Western Blot; METex14 = MET exon 14 skipping mutations; FISH = fluorescence in situ hybridization; DFS = disease-free survival; OS = overall survival.
HGF and MET OE have been studied in four retrospective studies that found that these factors could be associated with aggressive disease phenotype (high ki-67 score), more extensive pathological disease (pT and pN), and worse overall survival (OS) [
18,
19,
20,
21].
Three independent studies have shown that MET mRNA levels were associated with more extensive disease (pN) and worse disease-free survival (DFS) [
22,
23,
24].
Three studies reported that the MET gene copy number (GCN) in MET AMP, whatever the cut-off used, has an impact on survival [
25,
26,
27]. Beau Fowler et al. showed that MET AMP (not stating the cut-off) was associated with a worse OS only in adenocarcinoma (not statistically significant) [
25]. Okuda et al. showed that patients with stage II–IV and MET GCN > 3 copies had worse OS [
26]. Cappuzzo et al. documented that patients with MET GCN ≥ 5 copies were associated with aggressive disease (higher grade score), more extensive disease (pT) and worse OS [
27]. Instead, Park et al. failed to associate MET AMP (scored both through Colorado Criteria Cancer Center MET/CEP7 ratio ≥ 2 or Cappuzzo criteria GCN ≥ 5 copies) with clinical outcome, reporting that only MET OE was associated with worse OS [
28].
Only two Asian studies included all three MET parameters, MET OE, MET AMP, and MET exon 14 skipping mutations (METex14), in their analyses, stating that METex14 are extremely rare (less than 5%) and can be associated with worse OS [
29,
30].
3. MET as Predictive Biomarker
3.1. MET Exon 14 Alterations
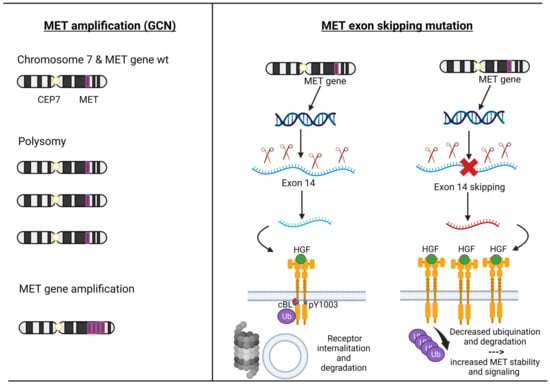
MET exon 14 encodes part of an important regulatory region and a binding site for cBL E3-ubiquitin ligase in the juxtamembrane domain of the MET receptor. Loss of this binding site leads to decreased ubiquitination, thereby reducing MET receptor internalization and degradation, resulting in increased MET levels [
31,
32,
33]. MET ex14 alterations comprised point mutation, insertion, and deletion. Most frequent are disrupted splice sites that lead to MET ex 14 skipping in the RNA transcript and a truncated MET receptor without the Y1003 ubiquitin ligase binding site [
34] (
Figure 2).
Figure 2. The pathogenesis of MET amplification and MET exon 14 alterations. Description: on the left, the mechanisms by which the MET gene can be amplified (polysomy versus intrachromosomal gene amplification) are depicted; the right shows the physiological mechanism of transcription and translation of the MET gene up to the degradation of the MET receptor and the pathological mechanism of the MET exon 14 skipping leading to the loss of Y1003, which decreased ubiquitination and degradation of the MET receptor. Abbreviations: MET = mesenchymal–epithelial transition; HGF = Hepatocyte Growth Factor; GCN = gene copy number; CEP7 = centromere of chromosome 7; wt = wild type. Created with
BioRender.com (accessed on 18 September 2023).
Frampton et al. analyzed tumour samples from 38,028 patients and found 221 (0.6%) cases with METex14: 3.0% in lung adenocarcinoma and 2.3% in non-adenocarcinoma lung cancers [
35]. Awad et al. reported the first retrospective study of METex14 NSCLC patients [
36]. Out of 993 non-squamous NSCLC patients, they identified 28 (3%) METex14 NSCLC patients: most of them had adenocarcinoma histology, while 14% of them had pleiomorphic lung carcinoma [
36]. In another retrospective series of 711 METex14 cases, 288 distinct METex14 mutations were identified [
37]; of these, 88% were ever smokers, and 12% were nonsmokers. According to the histology, 478 (67.0%) were non-squamous, 79 (11.1%) squamous, and 24 (3.2%) adenosquamous. The most common METex14 mutations were D1028H (8.1%), D1028N (7.8%), c.3082 + 2T.C (5.0%), D1028Y (4.6%), and c.3082 + 1G.T (4.4%) [
37].
An important issue is the diagnostic of METex14 mutations due to their relative rarity and complexity, as not all of them lead to an exon 14 deletion. Next-generation sequencing (NGS) is the best choice as a testing platform because it has the advantage of detecting other oncogenic drivers concurrently using one test performed on a single sample. Given the complexity of the MET exon 14 skipping mutations, different tools to target enrichment for NGS vary dramatically in their ability to detect these events [
38]. DNA-based amplicon NGS assays have a low detection rate of METex14 (around 63% of the cases) [
39]. The detection rate of METex14 can be improved by including fragment analysis and three additional amplicons to sequence exon 14 [
40]. The hybrid capture library (which uses longer pieces of DNA region with respect to the primers) with optimized bioinformatics tools can improve the detection rate with respect to DNA-based NGS [
41]. On the other hand, two reports have demonstrated that RNA-based NGS assays can be superior to DNA-based NGS tools in detecting METex14 [
42,
43]. However, one of the major caveats of RNA-based NGS is that RNA is more vulnerable to degradation than DNA, which leads to a reduction in the quality of RNA for formalin-fixed, paraffin-embedded samples [
44]. An optimal solution could be represented by DNA-/RNA-based NGS assays such as Illumina or Thermo Fisher platforms [
45,
46]. Sun et al. theorized a calculation system to hypothesize which METex14 mutations really lead to exon 14 deletion and can be useful, above all, for extremely rare or novel mutations [
47].
ESMO guidelines strongly recommend testing for METex14 skipping through an NGS panel. When using an RNA-based assay, it is mandatory that adequate internal validation and quality control measures are in place to ensure that rare gene mutations are not missing, and liquid biopsies can be used to test for molecular drivers. However, all patients with a negative blood test still require tissue biopsy [
48].
3.2. MET Amplification
The MET amplification (MET AMP) is due to an increase in the copy number of the MET gene (GCN), resulting in protein overexpression. True gene amplification results from gene duplication should not be confused with increased MET copy number due to high polysomy where there is complete chromosome duplication leading to multiple copies of chromosome 7 in tumour cells. Distinguishing between these two mechanisms is critical as true MET amplification is thought to drive oncogenesis, whereas polysomy usually does not [
33] (
Figure 2).
MET amplification (MET AMP) as a ‘druggable driver’ has a longer and more complex history because, in this circumstance, we need to select the real gene-addicted tumours to avoid the ‘dilution effect’ that consists of including a heterogeneity patient population that comprises polysomy and low gene amplification.
In fact, the frequency of MET GCN in NSCLC ranges from 0.7% to 21%, depending on the technique used and the cut-off point for positivity [
49]. Fluorescence in situ hybridization (FISH) was the first validated method for detecting high MET copy numbers and, to date, remains the gold standard method [
48].
One of the first cut-offs used was the University of Colorado Cancer Center (CCC) criteria, which established the gene amplification on the basis of GCNs and the ratio between GCN and centromere of chromosome 7 (MET/CEP7). It also distinguishes low polysomy (≥4 copies in 10–40% of cells), high polysomy (≥4 copies in ≥40% of cells), and gene amplification (presence of tight gene clusters and a MET/CEP7 ratio of ≥2 or 15≥ copies of MET per cell in ≥10% of cells) [
50]. The next cut-off was established by Cappuzzo et al., where MET GCN was classified into those with a mean greater than or equal to five copies per cell versus less than five copies. Using this system, they identified 48 (11.1%) patients out of 435 cases, where true amplification was identified in 18 (4.1%) patients [
27]. Subsequently, two studies used the Cappuzzo criteria and CCC criteria, finding MET+ cases in 7% and 4.2%, while the true amplification was only 2.4% and 1.1% [
28,
30].
Noonan et al. analyzed 1164 patients with adenocarcinoma with information on MET/CEP7 ratio and 686 with information on mean MET per cell value from two separate cohorts, and the GCN was reported by two methods: mean MET copy number per cell value (low ≥ 5 to <6 copies; intermediate ≥ 6 to <7 copies; high ≥ 7 copies) and the ratio MET/CEP7 (low ≥ 1.8 to ≤2.2; intermediate > 2.2 to <5; or high ≥ 5) [
51]. Out of 686 cases, 99 (14%) had a MET ≥ 5 copies (FISH+), and 52 of 1164 (4.5%) had a MET/CEP7 ratio ≥ 1.8, meanwhile only 4 (0.03%) patients had a high amplification (ratio ≥ 5). Overall, 61% of 1164 patients had concomitant gene mutations. According to the MET/CEP7 ratio, the concomitant gene mutations were distributed as the following: low (15 of 29, 52%), intermediate (9 of 18, 50%), and high (0 of 4, 0%,
p = 0.04). This rigorous cut-off (ratio > 5) helped to identify extremely rare true amplificated cases (less than 1%) without other concomitant gene alterations [
51]. However, this analysis was not conclusive, and even now, several cut-offs are being used.
It should be noted that MET AMP has been described in patients with NSCLC not previously exposed to tyrosine kinase inhibitors (TKIs) (de novo MET amplification) or as a mechanism of resistance to TKIs, mainly to EGFR (acquired MET amplification).
3.3. MET Overexpression
MET protein overexpression ranges from 15 to 70% in NSCLC [
28]. The MET receptor overexpression (MET OE) is measured by immunohistochemistry (IHC). It is found that there is a poor correlation with MET gene amplification or mutation. In the Lung Cancer Mutation Consortium multi-institutional cohort study of seventy-one IHC-positive cases, only one (1%) was MET AMP, and two (3%) were METex14-mutated; instead of the 110 MET IHC-negative cases, two (2%) were MET-amplified [
52]. In addition, several phase 3 trials with MET overexpression treated with MET-targeted therapies found that protein overexpression was an unreliable biomarker; thus, the utility of this biomarker is limited in clinical practice [
52].
4. Targeting MET
In the past, two randomized phase III trials with onartuzumab (a monoclonal antibody) and tivantinib (a selective MET TKI inhibitor) in the second/third line were conducted in an unselected NSCLC patient population, and these trials failed to meet their primary endpoint in improving the OS [
53,
54] (
Table S1).
Additionally, several MET-Is have been developed despite the failure of these first approaches [
53,
54]. MET-Is can be distinguished because of the mechanism of trapping MET: type I inhibitors compete with ATP to the ATP-binding pocket of the active conformation of MET, type Ia (crizotinib) interacts through the G1163 site, while type Ib (capmatinib, tepotinib, and savolitinib) interaction is independent of the G1163 site; type II ATP-competitive MET kinase inhibitors (cabozantinib, glesatinib, and merestinib) are defined by their ability to inhibit MET in its inactive state [
55,
56].
5. Resistance Mechanisms to MET-Is
5.1. Intrinsic Resistance
It refers to primary resistance and, to our knowledge, is a phenomenon poorly described for MET+ NSCLC treated with MET-I. Moreover, there are currently no translational studies that have investigated epigenetic alterations that could have helped us better understand the mechanisms of resistance to inhibitory METs. Type I MET inhibitors’ activity does not seem to be negatively influenced by the different types or localizations of the METex14 mutations [
68,
74,
84,
93]. Subgroup analyses for evaluable patients with sufficient tumour samples for NGS from PROFILE-1001 and phase II trial of savolitinib and NGS analyses from liquid biopsies from the VISION trial showed that patients with METex14 NSCLC could have concomitant gene mutations and that the most common are p53 (38–49%), MDM2 (20–25%), and CDKN2A (20%) [
68,
84,
93]. Moreover, post hoc analyses showed that p53 could negatively affect the response (savolitinib trial), and a trend has been documented for the lasting benefit of p53 (VISION and savolitinib trials) and POT-1 in few patients with sarcomatoid histologies (savolitinib trial) [
84,
93,
95]. Importantly, for patients with high MET AMP NSCLC, the absence of concomitant gene mutations was associated with higher ORR in PROFILE-1001 [
69].
5.2. Acquired Resistance
In METex14 or MET AMP NSCLC, patients treated with MET-Is acquired mutations in the tyrosine kinase domain (TKD) and can sustain resistance to these inhibitors. Importantly, D1228 and Y1230 seem to mediate resistance to type I inhibitors by disrupting drug binding, while L1195 and F1200 mutations seem to confer resistance to type II inhibitors [
96,
97,
98,
99,
100,
101,
102,
103,
104]. Moreover, an off-target mechanism of resistance to MET inhibitors involving KRAS gene amplification or mutations has been proved both in preclinical and clinical models [
105,
106]. One preclinical study with selected MET-Is showed that mutations within the MET activation loop (D1228N, Y1230C/H) were associated with resistance to type I MET-Is but remained sensitive to type II inhibitors (glesatinib) [
98].
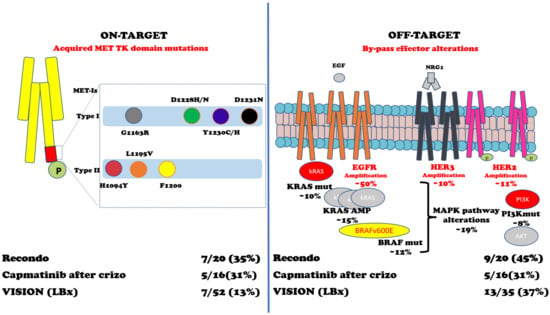
In a retrospective study, Recondo et al. reported 20 patients with METex14 NSCLC treated with MET-Is [
102]. They found genomic alterations (GAs) potentially related to resistance to MET-Is in 15 (75%) patients: 7 (35%) patients developed single or compound MET mutations on TKD in codons G1163R, D1228H/N, Y1230C/H/S, and L1195V (associated with type I inhibitors crizotinib and capmatinib) and H1094Y and L1195V (type II inhibitor glesatinib) plus high MET AMP, while 9 (45%) patients were associated with off-target alteration including KRAS (mutations or amplifications) and gene amplification of EGFR, HER3, and BRAF; one patient developed both on- and off-target mechanisms of resistance. It should be noted that two out of six patients (33%) were sensitive to the MET-I switch (the first case was initially treated with crizotinib and subsequently with merestinib; the second was treated with glesatinib and then with crizotinib) [
102].
A second Chinese retrospective study has been conducted with 86 patients with NSCLC harbouring MET TKD mutations [
103]. Importantly, they screened three different cohorts of patients (treatment-naïve patients with EGFR+ NSCLC treated with either EGFRTKI and/or MET inhibitors), and they found that ‘acquired’ MET mutations are extremely rare in treatment-naïve patients (0.06%) and have higher frequency after EGFR/MET-Is [
103].
From a small phase II trial of capmatinib in 20 patients with METex14 NSCLC after crizotinib treatment, among the sixteen patients with detectable ctDNA, five (31%) had acquired MET mutations, three (19%) had MAPK pathway alterations, and two (13%) had ERBB pathway alterations [
104]. From the VISION trial of tepotinib, 52 pts with progression had end-of-treatment liquid biopsy samples [
95]. Emerging MET resistance mutations (Y1230H/C and D1228H/N, plus three unknown functions G685E, G344R, and S156L mutations) occurred in seven (13%) patients and off-target mechanisms p53/RB1 mutations (6/35), EGFR/HER2 amplifications (4/35), and PI3K/RAS mutations (3/35) [
95]. Two independent studies have shown that KRAS alterations (amplification or mutations) can play a significant role in acquired resistance to MET inhibitors [
105,
106].
As for MET AMP NSCLC, nine evaluable patients out of twenty-four patients enrolled in cohort B (MET amplification) of the VISION trial have had liquid biopsy profiles at disease progression: two (22.2%) of them had acquired MET resistance mutations; one patient had D1228H/N/Y, Y1230C/H, and D1231N; and another had D1228N/H, Y1230H, and D1231N (90). Figure 3 and Table 2 summarize the intrinsic and acquired mechanisms of resistance to MET-Is.
Figure 3. Acquired mechanisms of resistance to MET-Is. Abbreviations: MET TK domain = MET tyrosine kinase domain; MET-Is = MET inhibitors; Crizo = crizotinib; EGF = Epidermal Growth Factor; NRG1 = neuregulin 1; EGFR = EGF receptor; HER3 = human EGFR-3; HER2 = human EGFR2; kRAS = Kirsten rat sarcoma virus gene; BRAF = v-raf murine sarcoma viral oncogene homolog B1 gene; MAPK = mitogen-activated protein kinases pathway; PI3K = Phosphatidylinositol 3-kinase; AKT = Ak strain transforming kinase protein; Mut = mutations; AMP = amplification. References: Recondo G et al. [
102]; Dagogo-Jack et al. [
104]; Paik K et al. [
95].
Table 2. Studies that reported resistance mechanisms to MET-Is.
Abbreviations: MET-Is = MET inhibitors; Type inh = type inhibitors; Retrosp. = retrospective; TK = tyrosine kinase; Tx naïve = treatment-naïve; pts = patients; EGFR+ = Epidermal growth factor receptor mutated; METex14 = MET exon 14 skipping mutations; MET AMP = MET amplification; Crizo = crizotinib; KRAS = Kirsten rat sarcoma virus oncogene; ERBB = EGFR family; HER2 = human epidermal growth factor receptor 2; PI3K = Phosphatidylinositol 3-kinase; MAPK = Mitogen-activated protein kinase.
6. MET and Immunotherapy
Pivotal clinical trials of ICI have excluded or not included patients with known MET alterations NSCLC; therefore, our knowledge of efficacy in this target patient population can only be derived from translational research and retrospective trials [
107]. MET OE NSCLCs were associated with higher PD-L1 expression and T-cell infiltration [
108].
MET AMP NSCLC correlated with higher PD-L1 score, CD8+ T-cell infiltration, higher incidence of concomitant gene mutations and worse OS [
109]. Patients with high MET AMP NSCLC (stated as >10 copies) experienced a statistically significant benefit on OS when treated with ICI added to chemotherapy [
110].
Instead, there are conflicting results regarding patients with METex14 NSCLC and PD-L1 expression: one study showed that they are associated with higher PD-L1 score [
111], another study documented higher CD8+ T-cell infiltration but no higher PD-L1 levels [
112], while another third study found that they did not have high median Tumour mutational burden (mTMB) nor high PD-L1 levels [
113].
Sabari et al. reported 24 patients with METex14 NSCLC treated with ICI; the ORR was 17%, and the mPFS was 1.9 months. Responses were not enriched in tumours with PD-L1 ≥ 50% nor high TMB [
113].
Dudnik et al. reported 14 patients with METex14 NSCLC and 5 patients with MET AMP (cut-off not specified) NSCLC [
114]. The activity of ICI was modest in both groups of patients: in the first, the ORR was 12% (none out of two patients responded to treatment among those with PD-L1 ≥ 50%) with an mPFS of 4 months (1.9 months in the two patients with PD-L1 ≥ 50%); in the latter, the ORR was 25% (one out of four patients included in the analysis) with an mPFS of 4.9 months [
114].
The IMMUNOTARGET registry was a large European multi-center retrospective trial also involving EGFR and KRAS and enrolled 551 patients with gene-addicted NSCLC [
115]. ICI was given mostly as the second- or third-line setting. Thirty-six patients had MET+ NSCLC (twenty-three patients with METex14 plus thirteen with MET AMP, cut-off not indicated), the median PD-L1 score was 30, and 76% of the patients were ever smokers. Among these patients, the ORR was 16%, the mPFS (principal endpoint of the study) was 3.4 months, and the OS was 18.4 months. The mPFS was not augmented, regardless of whether we considered the MET alteration subtype, the smoking status, or PD-L1 expression [
115].
The GFPC01-2018 study has enrolled 30 patients with METex14 NSCLC (enriched for PDL-1 positive and ever smokers) treated with nivolumab or pembrolizumab and showed an ORR of 36%, an mPFS of 4.9 months, and an OS of 13.4 months [
116].
This was recently reported in two retrospective studies. In the first study of 87 patients with METex14, NSCLC in the mPFS of ICI-based regimens was numerically low (2.4 months in patients with high PD-L1) [
117]. In the second study of 248 patients with METex14 NSCLC, the mPFS of ICI was inferior with respect to chemotherapy in the first-line setting (229 patients, 3.6 vs. 5 months) and second-line setting (158 patients, 3.3 vs. 3.9 months) [
118].
Post hoc analyses from GEOMETRY-mono1 and VISION trials have demonstrated that the activity of the MET inhibitor (capmatinib or tepotinib) was irrespective of prior therapies (including ICI); however, the median time to treatment of ICI in the VISION trial was less than 4.5 months, and the OS of patients treated with ICI alone was inferior respect to patients treated with chemotherapy +/− ICI (15.8 vs. 20 months) [
78,
119].