Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Obstetrics & Gynaecology
Hyperandrogenism encompasses a wide range of clinical manifestations, including polycystic ovary syndrome (PCOS), idiopathic hirsutism, hirsutism and hyperandrogaenemia, non-classical congenital adrenal hyperplasia, hyperandrogenism, insulin resistance, acanthosis nigricans (HAIR-AN), ovarian or adrenal androgen-secreting neoplasms, Cushing’s syndrome, and hyperprolactinaemia.
- hyperandrogenism
- endometrial receptivity
- androgen excess
- infertility
1. Introduction
1.1. Normal Androgen Physiology
Physiologically, androgens in women are produced in the ovaries, adrenal glands and peripheral tissues. Under normal circumstances, the ovaries and adrenal glands contribute approximately equally to testosterone production. Almost all testosterone is produced by direct testosterone secretion from the adrenal glands. A small portion of testosterone production is formed by peripheral conversion of circulating androstenedione, which is secreted into tissues mainly from the ovaries by the enzyme 17-β-hydroxysteroid dehydrogenase (HSD) [4].
Testosterone, dehydroepiandrosterone sulphate (DHEAS), dehydroepiandrosterone (DHEA), androstenedione, and androstenediol are the androgens released by the endocrine glands [5]. Androstenedione, which is synthesised in the ovaries and adrenal glands, is a direct precursor of testosterone [5]. DHEAS and DHEA are also precursors of testosterone [6]. DHEAS (produced in the adrenal glands) and DHEA (produced in both the ovaries and adrenal glands) are converted into androstenedione, which is then converted into testosterone. DHEAS is an essential measure of adrenal androgen production because it is produced exclusively in the adrenal glands [5]. These androgens are secreted in response to luteinising hormone (LH) in the ovaries and adrenocorticotropic hormone (ACTH) in the adrenal glands.
In addition to the ovaries and adrenal glands, androgens are also produced by the peripheral conversion of prohormones of adrenal and ovarian origin in non-endocrine tissues. In women, androstenedione is the predominant precursor of serum testosterone, making it the most important prohormone [7]. In addition, 5-alpha-reductase, found in the liver, hair follicles, and other androgenic target cells, convert testosterone into dihydrotestosterone (DHT), a potent androgen [8]. Figure 1 below shows a summary of the interconversion of testosterone and testosterone precursors [9].
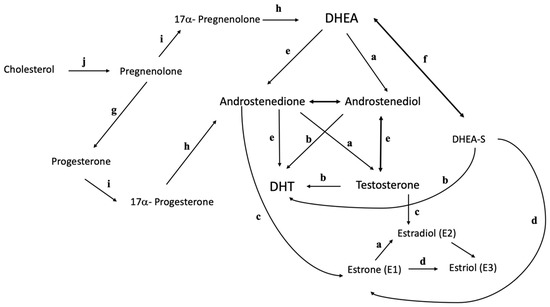
Figure 1. Interconversion of testosterone and testosterone precursors. a, 17-β-hydroxysteroid dehydrogenase; b, 5α-reductase; c, P450 aromatase; d, 16α- hydroxylase; e, 3β-hydroxysteroid dehydrogenase; f, DHEA sulfotransferase; g, 3β-hydroxysteroid dehydrogenase type 1; h, 17, 20 lyase; i, 17α-hydroxylase; j, cholesterol side chain cleavage.
Although the sites of androgen synthesis are clearly known, the regulation of androgen production is unclear. Currently, testosterone secretion is thought to be a by-product of oestrogen production in the ovaries and glucocorticoid production in the adrenal glands. It has been shown that androgen secretion in the adrenal glands is related to factors that influence cortisol secretion in addition to ACTH levels. Thus, an increase in cortisol and ACTH leads to an increase in serum androgens [7,10]. In contrast, the control of hypothalamic, pituitary, and ovarian hormones is more complex, with oestradiol and progesterone playing an important role in the regulation of gonadotropins.
Metabolic clearance of androgens also plays an important role in regulating serum testosterone levels. In addition to increased production of androgens, decreased androgen clearance results in increased serum androgen levels. Conditions associated with decreased androgen clearance include oestrogen treatment [4,11], barbiturate treatment [4,12], hyperthyroidism [4,13], hypogonadism [4,14], and ageing [15].
Besides oestrogen and progesterone, androgen also plays an important role in the menstrual cycle. During the oestrogen-dominated proliferative phase, stromal fibroblasts in the functional (upper) layer of the human endometrium exhibit the highest expression of AR [16], which is down-regulated during the secretory phase but is maintained in stromal cells within the basal compartment throughout the cycle [17,18]. Expression of AR is also increased in glandular epithelial cells and is up-regulated during the mid-secretory phase [19]. Furthermore, expression of AR has been detected in both stromal and epithelial cells in the first trimester decidua [19]. In contrast, AR has been identified in the perivascular stromal cells in the functional layer, but not in the endothelial cells [6].
In the context of endometrial receptivity, androgen receptor (AR) expression is restricted to the stroma of the endometrium and changes during the menstrual cycle, gradually decreasing from the early proliferative to the mid-secretory phase [20]. Regulation of uterine androgen receptors (AR) contributes to the normal pregnancy process. This reduction has been found to correlate with significant differential expressions of the Spp1, Prl, Igfbp1, and Hbegf genes that are associated with endometrial receptivity and endometrial decidualisation [21]. Hyperandrogenism leads to increased expression of AR and implantation failure due to aberrant expression of genes related to implantation and mitochondrial function.
1.2. Hyperandrogenism
The source of hyperandrogenism may be the ovaries and the adrenal glands. Typically, androgen secreted by the adrenal gland predominates in non-classical congenital adrenal hyperplasia (NC-CAH), while the ovary is the main source of androgens in polycystic ovary syndrome (PCOS) and idiopathic hyperandrogenism [22,23]. However, previous data suggest that 35% of PCOS cases, 50% of idiopathic hyperandrogenism cases, and about 70% of patients with NC-CAH have excess androgens from more than one source [24]. A more detailed explanation of the non-PCOS causes is discussed in the next section.
The primary source of androgens in PCOS, the most common disorder with androgen excess, is the ovaries. Ovarian hyperandrogenism is the primary pathogenetic mechanism for the syndrome, although no genetic impairment of ovarian enzymes has been identified [25]. In addition, both increased primary androgen secretion by the theca cells and increased drive by luteinising hormone (LH) (and insulin) contribute to increased ovarian androstenedione and testosterone production [26,27]. Theca cells from polycystic ovaries even produce more androstenedione both under basal conditions and during stimulation by gonadotrophins [26,27].
However, in many women with PCOS, there are multiple causes of androgen hypersecretion. About 50% of women with PCOS have elevated circulating levels of dehydroepiandrosterone sulphate (DHEAS) and 11β-hydroxyandrostenedione, two androgens secreted almost exclusively by the zona reticularis of the adrenal glands [28,29]. The elevated DHEAS levels in women with PCOS are likely due to the cumulative effect of several factors, including increased circulating unbound oestradiol levels and altered cortisol metabolism [30,31]. Indeed, decreased peripheral cortisol has been observed in PCOS, due to increased inactivation of this steroid by 5-alpha-reductase or impaired reactivation of cortisol from cortisone by 11beta-hydroxysteroid dehydrogenase type 1 [32,33]. Therefore, the decrease in peripheral cortisol would lead to decreased negative feedback on ACTH, so that the activity of the pituitary–adrenal–androgen axis would increase to maintain normal cortisol levels.
Elevated insulin levels are also thought to be the main cause of increased adrenal androgen secretion in women with PCOS [34]. However, serum DHEAS levels are typically lower in obese women with PCOS [35], and serum DHEAS correlates negatively with serum insulin in hyperandrogenic women [24]. Therefore, it is unlikely that hyperinsulinemia is the primary cause of elevated DHEAS levels. However, this does not rule out a role for insulin in adrenal androgen excess in women with PCOS.
In general, the source of androgen is not thought to affect the phenotype of diseases with androgen excess. While there are many differences in phenotype between the various androgen disorders, particularly between severe (PCOS) and mild syndromes [36], these differences appear to be determined primarily by other features of the syndrome (mainly insulin resistance). However, regardless of the type of androgen excess disorder (PCOS or idiopathic hyperandrogenism), hyperandrogenic patients with elevated DHEAS tend to be leaner, and have lower insulin levels and a better metabolic profile [36]. These findings have yet to be confirmed, but they raise the possibility that elevated DHEAS levels may have a protective effect on metabolic syndrome or that elevated insulin levels suppress DHEAS secretion [37].
2. Hyperandrogenic Syndromes
2.1. Androgen-Secreting Tumours
Pure androgen-secreting adrenal tumours were found less frequently compared to other adrenal tumours. Most adrenal tumours are clinically silent, but when functional they usually secrete cortisol, aldosterone, or catecholamines. This leads to the symptoms of androgen-secreting tumours, which can be divided into three categories: hirsutism, virilisation, and menstrual cycle disorders [15]. On the other hand, only about 1% of ovarian tumours secrete androgens, resulting in clinical hyperandrogenism. The most common androgen-secreting ovarian tumour is the Sertoli–Leydig cell tumour, which accounts for about 0.5% of all ovarian tumours. In women of reproductive age, this tumour is usually benign and unilateral.
Androgen-secreting tumours are a rare cause of hirsutism. In more than half of the reported cases, the tumours proved to be malignant and may be of ovarian or adrenal origin [38]. The typical presentation is hirsutism (excessive hair growth in androgen-sensitive areas such as the face, chest, nipples, buttocks, and external genitalia). Increasing musculature, a deeper voice, breast atrophy, male pattern baldness, and clitoromegaly are signs of virilisation. [15,38].
Physical examination may indicate abdominal or pelvic masses; if these originate from the adrenal gland, there is often concomitant hypercortisolaemia and elevated levels of dehydroepiandrosterone and dehydroepiandrosterone sulphate (DHEAS), leading to Cushing’s syndrome [38]. In a patient with suspected hyperandrogenism associated with an androgen-secreting tumour, a thorough examination should be performed to assess the severity, timing of onset, and progression of symptoms. In postmenopausal women with an unknown cause of increased testosterone and virilisation, the suspicion of an androgen-secreting tumour should be suspected, whether it is ovarian or adrenal in origin [39].
2.2. Congenital Adrenal Hyperplasia
Congenital adrenal hyperplasia (CAH) is inherited in an autosomal recessive manner and disrupts adrenal steroidogenesis [40,41]. It is caused by the absence of one of the enzymes involved in the synthesis of adrenal steroid hormones, usually a deficiency of 21-hydroxylase, which diverts the precursors into the androgen pathway [41,42]. The specific mutation of the genes involved in the disruption of adrenal steroidogenesis results in different clinical features. In 21-hydroxylase deficiency, the clinical spectrum ranges from salt-wasting to simple or mild virilisation.
In classical CAH (CCAH), features of both salt wasting and simple virilisation are observed [40]. The mild variant is called non-classical CAH (NC-CAH). CAH is characterised by cortisol deficiency, with or without aldosterone deficiency, and androgen excess [41]. Physiologically, the hypothalamic–pituitary–adrenal (HPA) axis regulates cortisol secretion from the adrenal cortex. Corticotrophin-releasing hormone (CRH) produced by the hypothalamus regulates the release of adrenocorticotrophic hormone (ACTH), which subsequently stimulates the adrenal cortex. When the adrenal gland releases cortisol, a negative feedback mechanism signals the hypothalamus to regulate the secretion of CRH and ACTH [42].
In CCAH, impaired cortisol synthesis leads to loss of negative feedback inhibition of cortisol, an increase in hypothalamic CRH, and an increase in ACTH secretion in the pituitary. The excessive ACTH secretion leads to an accumulation of cortisol. The hyperandrogenic element associated with these enzyme deficiency disorders is the result of altered metabolism of cortisol [40]. Decreased enzymatic activity of 21-hydroxylase impairs the biosynthesis of cortisol, leading to an increase in the concentration of 17-hydroxyprogesterone (17-OHP) and progesterone. This leads to poor cardiac function, increased secretion of antidiuretic hormone (ADH), and exacerbated mineralocorticoid deficiency in affected individuals [43].
In CAH, the concentration of the substrates in close proximity to 21-hydroxylase, progesterone and 17-OHP, is increased due to the deficiency of 21-hydroxylase. In addition, the defective proteins are confined to the adrenal cortex. The androgen receptor in the adrenal cortex affects steroid metabolism and response to steroids. Apart from this, an increase in the concentration of dihydrotestosterone, DHT (17-OHP, which is converted to DHT by 5α-reductase) from alternative pathways of steroidogenesis further aggravates the symptoms of androgen excess. This pathway is probably the cause of the prenatal virilisation seen in affected female foetuses due to androgen excess [40].
In affected infants, CCAH usually occurs in the neonatal period. Symptoms vary depending on the sex of the infants [40]. Diagnosis of the disease is difficult, especially in affected neonates, who are at higher risk of hyponatraemia, hypokalaemia, and hypotension with an unknown, possibly fatal, outcome [44]. Prenatal virilisation also makes it difficult to determine the “true” sex of the newborn at birth. In most cases, they have ambiguous genitalia [40]. Therefore, a higher index of suspicion for 21-OHD is required to be excluded in this case.
Children with NC-CAH often show premature puberty (premature development of pubic hair, axillary hair, or apocrine odour before the age of 8 or 9 in girls and boys, respectively). Girls may have the enlargement of the clitoris, while boys may have phallic enlargement with testes of prepubertal size. Although premature puberty is common in children with CAH, it is a rare cause of premature adrenarche [45]. CAH is diagnosed when testosterone, 17-OHP, and androstenedione are elevated, with or without advanced bone age [40,46].
Common symptoms in female adolescents and adults with NC-CAH include irregular menstruation, chronic anovulation, infertility, acne, and hirsutism, which is reported as the most common symptom [47,48,49,50]. Distinguishing between NC-CAH and polycystic ovary syndrome (PCOS) is quite difficult due to the similarity of symptoms [43,51]. It has been shown that women with NC-CAH are more likely to have elevated 17-OHP and progesterone levels compared to women with PCOS [52]. Women with PCOS usually have oligomenorrhoea, biochemical or clinical signs of hyperandrogenism, obesity, insulin resistance, polycystic ovarian morphology, and an increased LH/FSH ratio. However, even with these features, it is difficult to distinguish women suffering from NC-CAH from PCOS [53]. In a previous study, it was reported that the women on NC-CAH did not suffer from symptoms of androgen excess. They achieved normal body size without adrenal insufficiency and also suffered from infertility [54].
As mentioned earlier, women with NC-CAH have many similarities with PCOS individuals. One mechanism of PCOS is disruption of the hypothalamic-pituitary-ovarian (HPO) axis, which can also occur in individuals with NC-CAH. It has been postulated that androgen overexposure in the uterus leads to changes in the mechanisms that control GnRH and kisspeptin. In another study, it was found that an increase in LH concentrations with an increase in LH pulse amplitude was highly associated with high androgen concentrations in women with NC-CAH [55,56].
Ovarian function can be directly influenced by androgens. The occurrence of polycystic ovarian morphology is highly influenced by excessive androgens, which may arise either from endogenous androgens as in CAH or from exogenous androgens as in illicit steroid abuse in female-to-male transgender or female athletes [56,57]. Androgen receptors are expressed in granulosa cells, theca cells, and oocytes [58]. A study using tissue-specific androgen receptor knockout mice revealed that androgens regulate follicular growth via androgen receptors at different stages of follicular development [59,60]. Physiologically, the function of androgens is to promote the initial growth of small antral follicles. However, hyperandrogenism leads to follicular arrest and the inability to select the dominant follicle [61]. Androgens can stimulate the extracellular matrix, leading to stromal hyperplasia. Thus, disruption of the HPO axis by androgens can occur at multiple sites [62].
Excess circulating androgens and progesterone may impair HPO axis function and endometrial receptivity. Increased progesterone concentrations impair the quality of cervical mucus, accelerate endometrial maturation, reduce endometrial receptivity, decrease sperm penetration, and impair embryo implantation [63]. Therefore, to ensure ovulation, proliferation of endometrium and implantation of the embryo, adequate suppression of progesterone (<60 ng/dL) and plasma renin activity is required in both CCAH and NC-CAH. Fertility rates are higher when glucocorticoid therapy is administered [50].
2.3. Hyperandrogenic Insulin-Resistant Acanthosis Nigricans (HAIR-AN) Syndrome
Hyperandrogenic insulin-resistant acanthosis nigricans (HAIR-AN) is a subtype of PCOS characterised by high insulin resistance [64]. Obesity, genetic, and environmental variables are associated with the development of HAIR-AN. Diagnosis is predominantly clinical, with laboratory studies providing further support. HAIR-AN syndrome is observed in 1 to 3 percent of hyperandrogenic women [45]. When triggered by LH or HCG, ovarian stromal cells synthesise androgens, depending on the pathophysiology. It has also been found to increase the steroidogenic activity of these cells. The latter is an important factor in determining the extent of hirsutism. There may be a closer relationship between the extent of hirsutism and the degree of hyperandrogenism observed. Insulin-like growth factor-1 (IGF1), a proteic peptide with close similarity to insulin, has the same steroidogenesis capacity as the latter [65].
It has been reported that maternal history of metabolic syndrome and the presence of acanthosis nigricans may lead to insulin resistance and be responsible for hyperandrogenism and virilisation. The approach to reduce insulin resistance was lifestyle modification. Treatment to reduce ovarian hyperandrogenism, such as combined oestrogen and progestin pills, are used because of their antigonadal effect, which inhibits LH, leading to a reduction in ovarian androgens; their increase in sex hormone-binding globulin is also known to reduce bioavailable testosterone [65].
Genetic insulin resistance disorders of type A are characterised by hyperandrogenism, insulin resistance, and acanthosis nigricans (AN). Type B IR is caused by circulating antibodies directed against the insulin receptor and is associated with other autoimmune diseases [66,67]. The HAIR-AN syndrome occurs in 1 to 5% of young women with hyperandrogenism, mainly in young black African women. Because of its subtle symptoms, its prevalence may be underestimated. It is a rare syndrome that causes an unusual multisystem disorder in women and is misdiagnosed in many cases [66].
Excess circulating insulin leads to increased expression of insulin growth factor receptors (IGFR) on epidermal keratinocytes and melanocytes, leading to AN [68]. Circulating insulin can also activate ovarian stromal cells and granulosa cells to produce excess androgens, which are responsible for the symptoms of hyperandrogenism. In addition to AN, clinical examination reveals indicators of virilisation, such as hirsutism, android obesity, clitoral hypertrophy, muscle hypertrophy and increased desire. Other symptoms of hyperandrogenism, besides virilisation, are amenorrhoea, hypofertility or infertility, retention acne, and androgenetic alopecia [69,70].
2.4. Hirsutism
Hirsutism is the excessive development of terminal hair in females in a typically male pattern [36]. It is usually a symptom of high androgen levels. Hirsutism occurs in 5–15% of women and is often associated with a lower quality of life and severe psychological distress. Hirsutism should be distinguished from hypertrichosis, which is defined as increased development of vellus hair in a general, non-sex-specific distribution independent of androgens. However, hyperandrogenism may exacerbate the condition [49].
Hirsutism is a clinical diagnosis, and the prevalence varies according to the diagnostic criteria used. The modified Ferriman–Gallwey scoring system, which consists of nine androgen-sensitive body regions, is commonly used to diagnose hirsutism [71]. Cut-off scores vary by race and ethnicity: hirsutism is defined as a score of 8 or higher in black and white women in the United Kingdom and the United States, and a score of 9 or higher in Mediterranean women. Hispanic and Middle Eastern women have a score of 6 or higher; South American women have a score of 2 or higher, and Asian women have a score of 2 or higher. Scores between 15 and 25 indicate mild hirsutism, while scores above 25 indicate severe hirsutism. The subjective nature of this scoring method, and the fact that locally high scores or previous cosmetic treatments cannot be taken into account, are two of the limitations of this method. The Endocrine Society advocates the treatment of patient-relevant hirsutism, i.e., excessive hair growth in the genital area from which patients suffer [38].
Idiopathic hirsutism is a diagnosis of exclusion that occurs in approximately 10% of women with regular menstruation, normal ovarian morphology, and normal serum androgen levels. Data suggest that almost half of all women with mild hirsutism have “idiopathic hirsutism”.
This entry is adapted from the peer-reviewed paper 10.3390/ijms241512026
This entry is offline, you can click here to edit this entry!