Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Ophthalmology
Autosomal recessive Stargardt disease (STGD1) is an inherited retinal degenerative disease associated with a mutated ATP-binding cassette, subfamily A, member 4 (ABCA4) gene. STGD1 is the most common form of juvenile macular degeneration with onset in late childhood to early or middle adulthood and causes progressive, irreversible visual impairment and blindness.
- Stargardt disease
- ABCA4
- gene therapy
- visual cycle modulators
- stem cell therapy
- adeno-associated viral vectors
1. Introduction
Autosomal recessive Stargardt disease (STGD1, OMIM#248200) is an inherited macular dystrophy also known as fundus flavimaculatus (the variant with more widespread flecks and less macular involvement), associated with disease-causing genetic variants in the ATP-binding cassette, sub-family A, member 4 (ABCA4) gene [1]. It is one of the most common inherited retinal diseases with a prevalence estimated at 1 in 8000–10,000 [2]. The carrier frequency is as high as 1 in 20 in some populations. The disorder is characterized by progressive central vision loss and may be accompanied by peripheral vision loss in later stages or more severe cases of the disease [3,4]. STGD1 commonly manifests during childhood or early adulthood, and less frequently in later adulthood, when later onset confers a better visual prognosis with a slower rate of visual loss [5]. The classical clinical features include variable pigmentary changes and pisciform flecks in the macula progressing to a variable extent of chorioretinal atrophy, which depends on the severity of the underlying pathogenic ABCA4 variants, with more severe variants seen in childhood-onset STGD1 [4]. Over 2000 variants within the ABCA4 gene have been described [6]. Patients report decreased visual acuity, central scotomas, and color vision abnormalities, with variable degrees of nyctalopia and visual field constriction in later stages or more severe cases. Diagnosis involves ophthalmic evaluation, multimodal imaging, and genetic testing [7]. The ABCA4 gene encodes a transmembrane protein, a member of the ATP-binding cassette transporter superfamily, sub-family A, member 4, which plays an essential role in the retinoid recycling in the visual cycle and localizes to the disc membranes in the outer segments of rods and cones. Lack of ABCA4 protein leads to accumulation of visual cycle byproducts (lipofuscin) and dysfunction of the retinal pigment epithelium (RPE) and subsequent photoreceptor degeneration [8,9]. Although no cure currently exists, promising treatment strategies including genetic therapy, pharmacological treatments, and stem cell therapy, are being explored. Advancing our understanding of the underlying mechanisms and developing effective interventions are essential for managing this debilitating retinal disorder and preventing irreversible vision loss.
1.1. Clinical Presentation and Diagnostic Testing
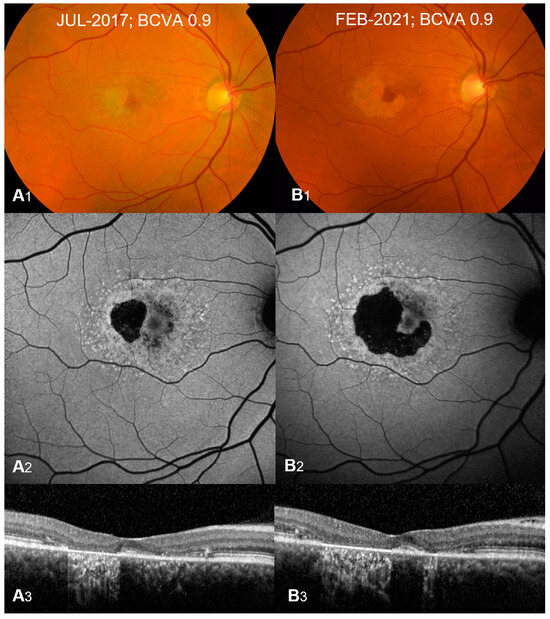
STGD1 presents with a spectrum of clinical features. On initial ophthalmoscopy, many children with STGD1 can have a normal-appearing fundus with or without vision loss; therefore, diagnosis can be delayed until the appearance of flecks or visual symptoms becomes significant enough to warrant deeper investigation with multimodal imaging including a combination of fundus fluorescein autofluorescence (FAF), optical coherence tomography (OCT), and electroretinography (ERG) [4,10,11,12,13]. Very young children may also present with symmetrical yellowish fine dots in the central macula. Later on, the characteristic key clinical features appear and include bilateral, progressive central vision loss, which encompasses both dyschromatopsia and central scotomata, macular atrophy, and yellow-white flecks within the RPE at the posterior pole [3]. The yellow-white flecks (Figure 1) represent the accumulation of lipofuscin, a yellowish autofluorescent pigment in the RPE cells that blocks the underlying choroidal fluorescence and causes the classic dark appearance of the choroid on FAF in 65–86% of patients [10,11].
Figure 1. Typical presentation of Stargardt Type 1 at diagnosis (A1–A3) and 3-year follow-up (B1–B3). Color fundus (A1,B1) shows macular atrophy with yellow-white retinal flecks. Fundus autofluorescence (A2,B2) shows patches of hypoautofluorescence surrounded by an increased signal with flecks of both increased and decreased autofluorescence. Optical coherence tomography B-scans (A3,B3) show outer retinal and retinal pigment epithelium loss with hypertransmission defects corresponding to atrophy. Atrophy growth is significant at 3-year follow-up (B1–B3) compared to presentation at diagnosis (A1–A3). Best corrected visual acuity (BCVA) remains stable at 0.9 Snellen decimals (20/160). Reprinted from [1], used under an open-access license agreement distributed under the terms of the Creative Commons CC-BY license.
FAF is a useful non-invasive imaging modality. It may show an increased signal from excessive lipofuscin and highlights the flecks with both increased and decreased autofluorescence in some patients and reduced central autofluorescence surrounded by a brighter signal similar to a bull’s eye appearance [1,10] (Figure 1). Areas of a decreased FAF signal may appear later and correspond to areas of RPE atrophy with secondary photoreceptor loss. A significant correlation has been shown between FAF subtype and genotype, with milder pathological variants producing a localized low signal at the fovea with a surrounding homogeneous background, whereas more severe variants (including nonsense) correlate with areas of hypofluorescent atrophic areas [10].
OCT typically shows outer retinal loss in the central macula (Figure 1) with possible peripapillary sparing of the retina and RPE, which if present strongly supports the diagnosis of autosomal recessive STGD1 and may also show the earliest abnormality in young children consisting of external limiting membrane thickening [10,12,14].
1.2. Genetics
STGD1 is caused by an autosomal recessive mutation of ABCA4 gene, located in the rim of outer segments of the photoreceptor cells and involved in the visual cycle. Photoreceptors during the visual cycle are constantly producing new disc membranes, pushing the older membranes further out towards the RPE [16]. One of the roles of the RPE is to phagocytose the most distal disc membranes with retinoid waste derivatives. ABCA4 proteins are “flippase” transporters, located in the outer segment disc membranes of rod and cone photoreceptors, which move retinoids from the lumen to the cytoplasmic side of the disc membranes mainly in the form of N-11-cis-retinylidene-phosphatidylethanolamine (NrPE) [8,17]. This allows the continuation of the visual cycle and decreases the production of bisretinoid compounds such as phosphatidyl-pyridium bisretinoid (A2PE) in the photoreceptor outer segment. As new disc membranes are produced, the older ones assume a more distal position, and the distal discs are phagocytosed by the RPE cells. After phagocytosis, the A2PE is converted to N-retinylidene-N retinylethanolamine (A2E) compound, which is insoluble and accumulates in the RPE cells to form lipofuscin (Figure 2) if it is not regularly managed by ABCA4 and other mechanisms [16]. With a mutated ABCA4, the accumulation of A2E and other bisretinoids leads to accumulation of toxic lipofuscin within the RPE, which with time will cause RPE cell death and photoreceptor dysfunction from a lack of RPE support [8,17]. The severity of phenotype correlates with the remaining ABCA4 function where more severe mutations, such as two null variants, result in a more severe phenotype with earlier onset and worse visual prognosis [13].
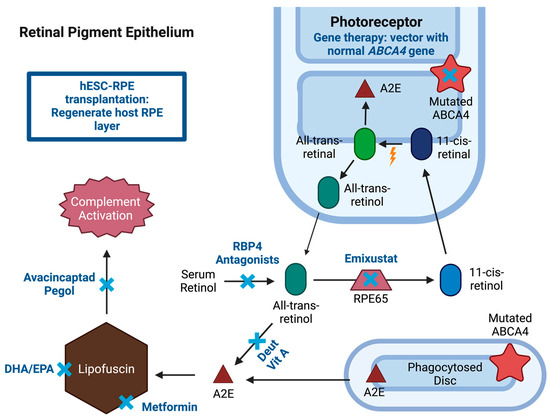
Figure 2. Potential pharmaceutical interventions for Stargardt disease (STGD1). Treatment options (blue “x”) are shown in a summarized representation of the visual cycle with mutated ATP-binding cassette, subfamily A, member 4 (ABCA4) gene resulting in the accumulation of lipofuscin in the retinal pigment epithelium (RPE). Treatment for STGD1 includes several strategies. Gene therapy includes vector delivery of human ABCA4 gene. Pharmaceutical therapies include visual cycle modulators (VCM), metformin, avacincaptad pegol, and docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA). VCM represented are retinol binding protein 4 (RBP4) antagonists, deuterated vitamin A, and retinal pigment epithelium-specific 65 kDa protein (RPE65) inhibitor (emixustat). Stem cell therapy includes human pluripotent stem cell-derived retinal pigment epithelium (hESC-RPE) transplantation for regenerating RPE layer. Created with BioRender.com.
2.3. Adeno-Associated Viral Vectors
AAV is a gene therapy delivery system that has shown safety with minimal adverse events in various trials; however, a major limitation of AAVs as vectors is their limited cargo capacity which is estimated to be around 4.7 kb [26]. Given the large coding region of the ABCA4 gene (50 exons and 150 kb), several in vitro models have been studied. The first attempt included creating an “oversized” transgene with the complete 6.8kb coding sequence yet transduction of these genes did not lead to the production of full-length protein [27]. Another model includes dual vector AAV strategies which can include fusing overlapping gene regions to create a transgene by fragmenting the original gene size. An alternative technique involves trans-splicing which provides no overlap between two transgenes since repeated sequences are removed, leaving an intact full-length coding sequence. Pre-clinical studies have shown early signs of success in delivering full-length ABCA4 coding regions using dual AAV and trans-splicing techniques (Figure 2) [28,29]. Given the positive outcomes obtained from animal studies, several studies are forthcoming using AAV as a gene therapy delivery system for individuals with STGD1.
AB0-504 (Abeona Therapeutics) is a dual AAV vector delivery system created by the Cre-LoxP recombinase system to reconstitute the full-length ABCA4 gene and is currently being tested in pre-clinical models. Preliminary results presented at the 26th Annual Meeting of the American Society of Gene & Cell Therapy provided evidence of the dual AAV vector system’s capability to generate the complete ABCA4 protein in cell culture [30]. Recent proof-of-concept investigations have further expanded upon these results, showcasing the expression of both ABCA4 messenger RNA (mRNA) and intact ABCA4 protein within the retinal tissue of ABCA4−/− knockout mice following subretinal administration. Notably, the observed levels of ABCA4 mRNA and full-length protein in the treated mice closely resemble those found in the naturally occurring ABCA4 of wild-type animals [30,31].
Moreover, other companies such as ViGeneron have also tapped into a novel dual AAV technology platform, achieving efficient expression of large genes in photoreceptors after intravitreal injection. They created VG-801 by using REconstitution Via mRNA Trans-splicing (REVeRT) technology enabling split genes encoding the 5′ and 3′ portions of human ABCA4 to be packaged into dual vgAAV vectors, which led to the generation of full-length protein. Notably, the REVeRT technology demonstrated high efficiency in reconstituting both transcript and protein levels of ABCA4 [32].
2.4. Optogenetics
Other companies are exploring the capability of AAV vectors to deliver genes that confer the inner retinal sensitivity to light bypassing degenerated photoreceptors altogether in end-stage Stargardt disease. In 2022, Nanoscope Therapeutics initiated a nonrandomized phase I/IIa clinical trial (NCT04919473) aimed at evaluating the safety and beginning to explore the efficacy of an intravitreal injection of an AAV viral vector carrying a multi-characteristic opsin-I (vMCO-I) gene which encodes a light-sensitive ion channel in patients with Stargardt disease due to ABCA4 or other genes. The vector leads to the expression of the opsin in bipolar cells. When illuminated by ambient light, it leads to depolarization of bipolar cells conferring sensitivity to ambient light. An ongoing multicenter open-label phase II clinical trial (STARLIGHT, NCT05417126) is being conducted to evaluate the safety and efficacy of a single intravitreal injection of vMCO-010 in up to six subjects with STGD due to ABCA4, ELOVL4, or PROM1 mutations and visual acuity worse than 20/640. The company recently reported on preliminary results at the 2023 American Society of Retina Specialists meeting showing 3 dB gain in mean visual field perimetry and no serious adverse events [37,38].
2.5. Non-Viral Delivery via Covalently Closed and Circular DNA (C3DNA)
C3DNA is a non-viral gene therapy platform developed by Intergalactic Therapeutics in IG-002 that enables the delivery of large genes, does not integrate into the genome, allows for redosing, eliminates safety concerns associated with immune reactions to viral vectors, and allows for precise engineering using synthetic biology [39]. The lead delivery modality for C3DNA therapies is cellular delivery of genetic material by Electro-Transfer (COMET) which combines a novel electrode design, custom wavelengths and algorithms, and an electrical field [40]. C3DNA does not use viral or bacterial sequences avoiding triggering immune reactions and potentially allowing redosing [40].
3. Pharmacological Therapies
3.1. Introduction
Emerging pharmacological therapies aim to reduce symptoms and inhibit the progression of STGD1 by targeting specific steps in the visual cycle that are altered by disease pathophysiology [42,43]. The visual cycle is a complex series of biochemical reactions that occur in the RPE and photoreceptor outer segments as part of the visual cycle, involving the primary photoreceptor molecule of vision composed of an opsin molecule and the chromophore 11-cis-retinal [44,45]. With light stimulation of the chromophore, 11-cis-retinal converts to all-trans-retinal, leading to the signal transduction cascade that allows the sensation of light [44,45]. The all-trans-retinal is then converted back to 11-cis-retinal through multiple steps in the outer segments and RPE cells involving numerous enzymes, one of which is the retinal pigment epithelium-specific 61 kDa protein (RPE65) [46]. ABCA4 transporters, found in the photoceptor outer segment disc membranes, are necessary for the elimination of all-trans-retinal from the disc lumens and recycling back to 11-cis-retinal. With ABCA4 gene mutations found in STGD1, a nonfunctional ABCA4 protein hinders this removal of all-trans-retinal from photoreceptors, which leads to the production of A2PE within outer segments and buildup of A2E, a major component of toxic lipofuscin, in the RPE cells. This leads to degeneration of RPE cells and subsequent photoreceptor loss [16,17].
3.2. RPE65 Enzyme Inhibition
Emixustat hydrochloride (ACU-4429, Kubota Vision Inc.) is an orally available visual cycle modulator that binds the RPE65 isomerase (Figure 2). Inhibiting the RPE65 enzyme depletes regeneration of visual chromophore 11-cis-retinal, which in turn reduces the production of all-trans-retinal [47]. Since these substrates are necessary for the production of bisretinoid components including A2E, this can reduce the accumulation of lipofuscin toxins in the RPE and decrease the rate of RPE and photoreceptor dysfunction and death [43]. In ABCA4−/− mice with A2E accumulation in RPE, treatment with emixustat significantly reduced A2E accumulation when compared to controls [48].
Phase 1 clinical trials (NCT00703183 and NCT00942240) with healthy subjects showed that once-daily oral administration of emixustat was able to slow the rod visual cycle and was well tolerated, even in multiple doses up to 75 mg, with dose-dependent inhibition of b-wave rod responses on ERG [42]. The drug was originally developed for slowing the progression of geographic atrophy (GA) in age-related macular degeneration (AMD) but failed to show a significant difference in lesion growth rate for treatment compared to the placebo in phase 2 (NCT01002950) and phase 2b/3 (NCT01802866) clinical trials [49,50]. It is currently being assessed as a potential treatment for STGD1. A phase 2 trial (NCT03033108) was completed in 2017 in the US with 23 subjects treated with several doses of the drug for 1 month, which demonstrated emixustat biological activity with suppression of the visual cycle in STDG1 patients. This was shown with suppression of recovery in b-wave amplitude after photobleaching in rods. Rod b-wave amplitude of ERG represents the extent of rhodopsin regeneration with a proportional relationship between the magnitude of rod–wave amplitude and rhodopsin levels. The rate of recovery over time reflects the regeneration rate of rhodopsin levels and therefore 11-cis-retinal levels, which represents RPE65 activity [43]. Near complete suppression of b-wave amplitude recovery post-photobleaching was reported at 10 mg of emixustat. Few clinically significant findings were observed with a BCVA change from baseline of −11 to +9 letters on the Early Treatment Diabetic Retinopathy Study (ETDRS) VA chart. The main ocular side effects were delayed dark adaptation and dyschromatopsia with no serious adverse events reported [43].
3.3. Vitamin A Dimerization Inhibition
ALK-001 (Alkeus Pharmaceuticals) is a deuterated vitamin A molecule that inhibits vitamin A dimerization. In the visual cycle without functional ABCA4 protein, vitamin A derivative all-trans-retinal converts to toxic bisretinoids such as A2E found in lipofuscin (Figure 2) [52]. Accumulation of lipofuscin leads to RPE and photoreceptor degeneration in STGD1 [52]. Decreasing vitamin A levels has been shown to slow A2E biosynthesis and considered a potential therapeutic target. A deuterated vitamin A has C20 hydrogen atoms replaced with deuterium atoms forming C20–D3-Vitamin A. Unlike the C20 carbon–hydrogen bond, the C20–D3 bond is much more difficult to cleave, which inhibits vitamin A dimerization, limiting production of A2E [52]. In ABCA4−/− mice, a diet containing C20–D3 compared to a diet with vitamin A led to reduction in A2E levels, lipofuscin deposition, and improved ERG, indicating improved eye function [53]. Dietary C20–D3 in ABCA4−/− mice was also able to prevent development of disease phenotype with interruption of C20–D3 in diet leading to returned development of disease phenotype [54].
3.4. RPB4 Antagonists
Retinol-binding protein 4 (RPB4) antagonists are being explored as potential therapeutic agents for STGD1. RPB4 has been shown to be involved in STGD1 and dry AMD pathophysiology through the toxicity of accumulated lipofuscin in the RPE [56,57]. RPB4 is the only plasma transport that carries retinol (vitamin A) to peripheral tissues from the liver. In particular, it carries retinol to the RPE, leading to the formation of bisretinoids. A2E is a bisretinoid component, which is associated with the toxicity of lipofuscin [56,58]. Therefore, reducing RBP4 levels has been explored as a therapy for managing STGD1 by modulating the visual cycle and reducing bisretinoid synthesis.
Fenretinide (Sirion Therapeutics) is a synthetic retinoid RPB4 antagonist, which binds to RPB4, preventing the binding of retinol and leading to the elimination of the RBP4–fenretinide complex and decreased retinol concentration in plasma and the eye [59].
A1120 (ICR-14967, Stargazer Pharmaceuticals), STG-001 (Stargazer Pharmaceuticals), and tinlarebant (LBS-008, Belite Bio Inc.) are non-retinoid RPB4 antagonists. In ABCA4−/− mice, A1120 reduced serum RBP4 and accumulation of lipofuscin bisretinoids; however, it did not suppress b-wave ERG recovery after photobleaching [56]. This may indicate that A1120 may not be able to produce sufficient biological activity with adequate suppression of the visual cycle in STGD1 patients [56]. Unlike fenretinide, A1120 is not a retinoic acid receptor-alpha agonist, which may improve safety and reduce adverse effects [56]. Clinical trials will be needed to determine A1120 efficacy in STGD1. For STG-001, a phase 1 trial (ACTRN12619000816156) for safety and tolerability was completed in 2019 in healthy patients. A subsequent phase 2 trial (NCT04489511) was completed in 2021 for the safety of once-daily oral STG-001 for 28 days in 10 subjects with STGD1 disease with no reported serious adverse events despite some reported visual disturbances (Table 1B).
3.5. Complement C5 Inhibition
Avacincaptad pegol (Zimura, Inveric Bio, Granbury, NJ, USA) is a complement factor C5 inhibitor. Lipofuscin accumulation with increased bisretinoids including A2E in RPE can lead to complement activation (Figure 2) [63]. C5 complement is essential for the formation of an inflammasome and membrane attack complex in the final stage of the complement pathway, leading to cell death and activation of inflammatory responses [64]. Markers of complement activation were elevated in ABCA4−/− mice, which led to basal laminar deposits within Bruch’s membrane due to lipofuscin accumulation and photoreceptor degeneration [65].
Avacincaptad pegol is delivered by intravitreal injection and has been explored as a potential therapy for several disorders including STGD1, dry AMD, neovascular AMD, and idiopathic polypoidal choroidal vasculopathy (NCT03364153, NCT02686658, NCT03362190, NCT03374670). A randomized, double-masked phase 2/3 clinical trial (NCT02686658) was completed in 2019 with 286 subjects with geographic atrophy receiving intravitreal avacincaptad pegol. A significant reduction of 27% in geographic atrophy mean growth rate was observed in patients receiving either 2 or 4 mg doses versus sham over 12 months without serious adverse events [64]. Continued reductions in GA growth rate compared to sham were observed after 18 months in 2 mg and 4 mg cohorts, of 28% and 30%, respectively [66].
3.6. RPE Macroautophagy Stimulation
Metformin hydrochloride is a biguanide antidiabetic agent and is commonly prescribed oral medication for managing type 2 diabetes mellitus through suppressing liver gluconeogenesis and increasing peripheral insulin sensitivity (Figure 2) [67]. Metformin has also been reported to increase macroautophagy, essential for the degradation of cellular components damaged by reactive oxygen species, via the AMP-activated protein kinase (AMPK)-mammalian target of rapamycin (mTOR) pathway in many tissues [68]. AMPK maintains cellular energy homeostasis by regulating important processes including autophagy and can activate autophagy by inhibiting mTOR [69]. Interventions that increase stimulation of this pathway may improve RPE clearance of lipofuscin, which has the potential to decrease ABCA4 retinopathy disease progression by decreasing lipofuscin accumulation, thus slowing RPE and photoreceptor degeneration [70,71]. Metformin was shown to enhance autophagy via the AMPK pathway in human retinal pigment epithelial cells in response to hydrogen peroxide-induced oxidative damage [69]. In ABCA4−/− mice, oral metformin was shown to decrease lipid and lipofuscin accumulation in the RPE/choroid [72].
3.7. Omega-3 Fatty Acids
Docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), types of omega-3 polyunsaturated fatty acids, have been investigated due to their role as components of photoreceptor cells. These polyunsaturated fatty acids are esterified into phospholipids and help maintain photoreceptor membrane fluidity, retinal integrity, and visual function [73,74,75]. DHA is mainly located in photoreceptor cells and plays an essential role in the synthesis of disc membranes, rhodopsin activation, and rod and cone development. Supplementation of dietary DHA significantly increases DHA in the brain and retina [76]. Omega-3 fatty acids in preclinical studies have shown the potential to generate anti-inflammatory mediators and resolvins and reduce complement 3 and lipofuscin levels (Figure 2) [73,77,78].
4. Stem Cell Therapy
4.1. Introduction
Several stem cell studies in animals and humans have been conducted or are currently being conducted to investigate stem cell therapy for STGD1 in an effort to repair retinal tissue and consequently improve VA. These trials mainly focus on the use of human embryonic stem cells (hESCs) [82].
4.2. Human Pluripotent Stem Cell-Based RPE
Preclinical studies that transplanted hESC-derived mature RPE cells into the subretinal space of eyes in the rat model of inherited retinal degeneration, which undergo visual deterioration due to a mutation in Mertk (a c-mer proto-oncogene kinase receptor specific to RPE cells), showed promising results [83,84]. Rats that received a subretinal injection of 5000 to 100,000 hESC-RPE cells with concomitant immunosuppression had significantly better VA by optomotor responses compared to untreated animals without teratoma production or other adverse events [84]. The success achieved in preclinical studies led to the development of several phase I/II clinical trials for STGD1 worldwide (NCT01345006, NCT01469832, NCT02445612, NCT01625559, NCT02941991, NCT02749734, and NCT02903576). Trials with reported findings are summarized (Figure 2).
This entry is adapted from the peer-reviewed paper 10.3390/jcm12196229
This entry is offline, you can click here to edit this entry!