Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Pharmacology & Pharmacy
Cyclodextrins (CDs) are cyclic oligosaccharides bearing several glucopyranose residues connected by α-1,4 glycosidic bonds. Natural cyclodextrins contain six, seven, or eight glucopyranose units (α-, β- and γ-cyclodextrin, respectively) and are natural products, biodegradable, and generally lacking in toxicity. Cyclodextrins have toroidal shapes, with the smaller opening of the toroid (primary rim) corresponding to the C6-OH primary hydroxyls and the larger opening (secondary rim) to the C2-OH and C3-OH secondary hydroxyls.
- drug–cyclodextrin inclusion complexes
- nanosponges
- cyclodextrin nanofibers
- bioavailability enhancement
- cyclodextrin synthesis
1. Introduction
The functionalization of natural cyclodextrins is very important for pharmaceutical applications. Although natural CDs are hydrophilic, their aqueous solubility is relatively low due to their tendency to generate aggregates by intermolecular hydrogen bonding. This may cause some nephrotoxicity, especially in the case of β-CD, due to the renal accumulation of insoluble cyclodextrin crystals or cyclodextrin–cholesterol complexes. Thus, much effort has been devoted to the chemical modification of natural cyclodextrins in order to improve their solubility and complexation capacity. Over 11,000 cyclodextrin derivatives have been prepared by incorporation of various moieties such as amines, amino acids, peptides, and aromatic rings. Moreover, more sophisticated supramolecular systems such as polymers, metal–organic frameworks, hydrogels, and other supramolecular assemblies have also been assembled from cyclodextrins [1].
The chemical modification of cyclodextrins is carried out via reactions in nucleophilic hydroxyl groups. However, selective cyclodextrin functionalization, as opposed to random substitution, is a challenging task because their hydroxyl groups exhibit only slight differences in their reactivity. There are, nevertheless, some characteristic properties for each position that may allow for selective modification if moderately reactive reagents are employed: the hydroxyl group at position 2 is the most acidic, the hydroxyl at position 6 is the most nucleophilic, and the hydroxyl residue at position 3 is the least accessible due to steric hindrance and hydrogen bonding, and hence the least reactive [2]. On the other hand, cyclodextrins can form complexes with reagents in different ways depending on the size of the cavity and the solvent, modifying their reactivity and sometimes making their performance unpredictable [3] (Figure 1A). To add an additional layer of complexity, many different functionalization patterns can be obtained for the cyclodextrins resulting from a functionalization reaction (Figure 1B).
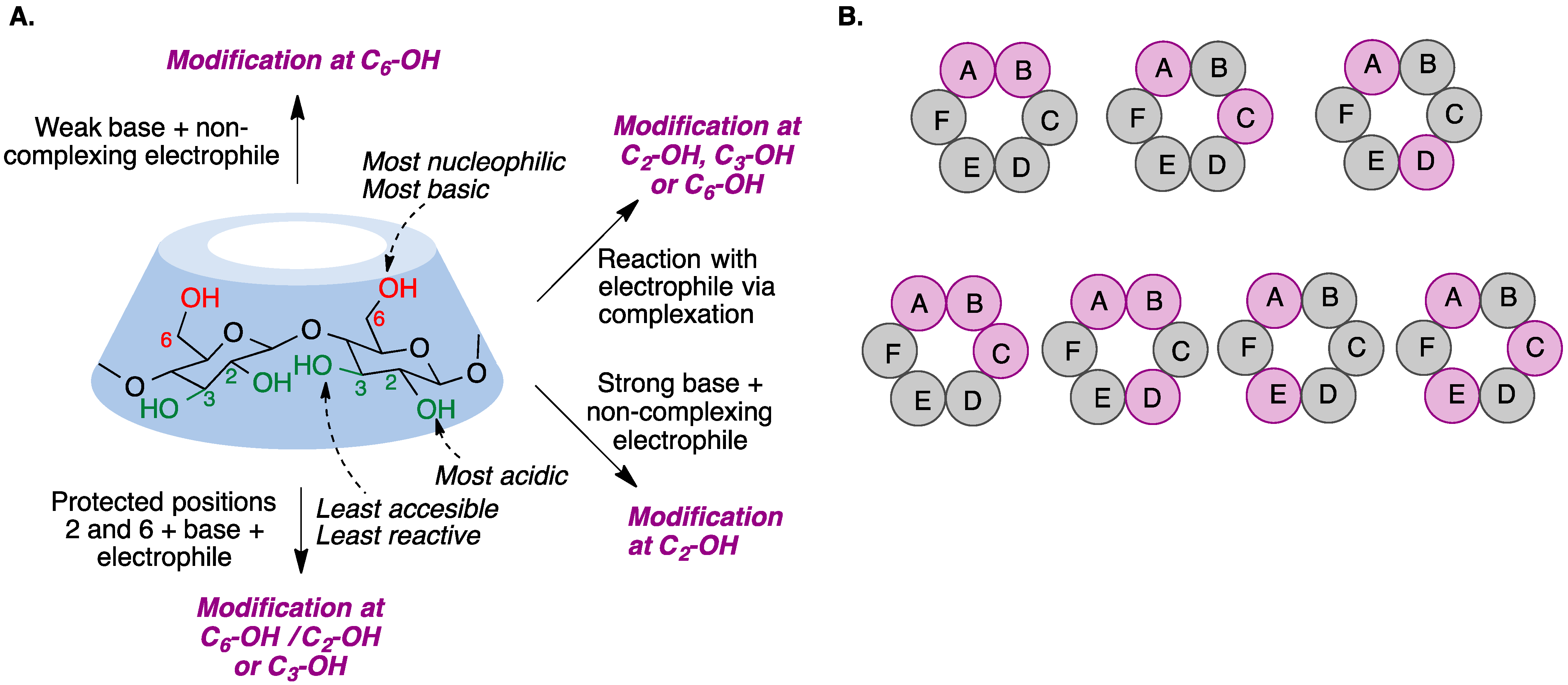
Figure 1. (A) Basic functionalization rules for cyclodextrins. (B) Positional isomers of difunctionalized (upper row) and trifunctionalized (bottom row) derivatives of α-cyclodextrin. The capital letters represent the individual glucose units.
Three main strategies can be employed for the preparation of functionalized cyclodextrins [4]:
- (a)
-
Straightforward selective modification of the desired positions. This is chemically challenging due to the competition between the hydroxyl groups at positions 2, 3, and 6 of the glucopyranose.
- (b)
-
Protection–deprotection strategies which involve taking advantage of the different reactivity of the hydroxyl groups toward protection and deprotection conditions. Deprotected hydroxyl groups can then undergo the desired modifications.
- (c)
-
Non-selective modification of hydroxyl groups to provide a mixture of derivatives that is then purified to isolate the desired one. This method generates a large number of undesired by-products, requires tedious purifications, and is not chemically efficient.
2. Selective Modification of the Primary Rim
2.1. Monosubstitution at C6-OH
Primary hydroxyl groups are more nucleophilic than their secondary counterparts, making modification of the primary rim of cyclodextrins relatively straightforward. Electrophiles react with the hydroxyl groups at C-6, yielding the desired product and an acid, which is neutralized by a base added to the reaction medium in order to avoid decomposition of acid-sensitive cyclodextrins. Common strong electrophiles suitable for nucleophilic attack by C6-OH include alkyl, sulfonyl, phosphoryl, silyl, and acyl chlorides [3][5]. Some representative transformations are summarized in Figure 2, and will be discussed below.
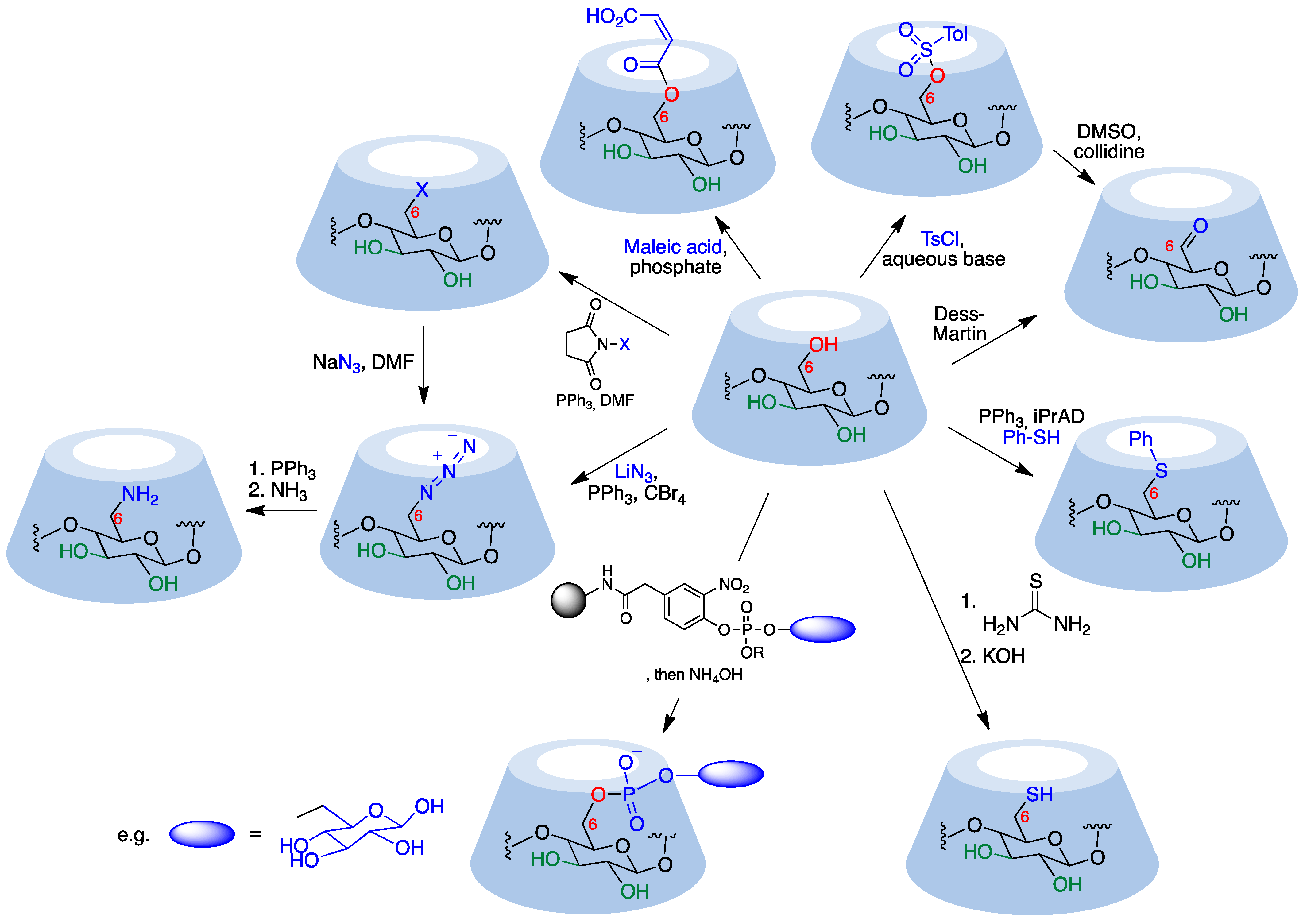
Figure 2. Representative monofunctionalization reactions at the cyclodextrin primary rim.
Monosubstituted O-sulfonylcyclodextrins are commonly used as substrates for the attack of nucleophiles to introduce functional groups at position 6. Mono-6-tosylcyclodextrin, for instance, is prepared via the reaction of the starting cyclodextrin with tosyl chloride in an aqueous basic environment [6][7] and can be further attacked by nucleophiles to yield 6-monofunctionalized cyclodextrins after chromatographic purification. Preactivation of the tosylate with imidazole improves selectivity toward 6-O-monotosylation [8]. The reaction with p-toluenesulfonic anhydride, on the other hand, does not require purification via chromatography since unreacted anhydride can be simply removed using filtration [9]. Strong bases need to be avoided because they can deprotonate the more acidic hydroxyl group in position 3, generating an alkoxide that displaces the leaving group at C-6 yielding a bicyclic structure.
Aldehydes are versatile groups for further functionalization. α, β, and γ-cyclodextrins can be converted to their mono-6-formyl derivatives in a straightforward manner using the Dess–Martin periodinane reagent [10]. An alternative, indirect way to obtain 6-formylcyclodextrins involves the reaction of the corresponding O-tosyl derivatives with DMSO and collidine [7][11].
The direct introduction of a single aromatic thioether is achieved via a thio-Mitsunobu reaction where β-cyclodextrin is mixed with thiophenol and diisopropyl azodicarboxylate in dimethylacetamide to yield the mono-6-phenylthio derivative with relatively good selectivity over the di- and trisubstituted by-products. The relative amounts of differently functionalized cyclodextrins could be tuned by modification of the reaction conditions [12].
A solid phase method provides mono-6-functionalized α and β-cyclodextrins bearing a variety of complex substituents bridged through a phosphodiester link from the unprotected native cyclodextrins. The Novagel amino solid support was derivatized with 4-hydroxy-3-nitrophenylacetic acid, which was then functionalized with a suitable phosphoramidite derivative containing the structural fragment to be attached to the cyclodextrin (e.g., a saccharide). Cyclodextrins were added to this support in the presence of 1-(mesitylene-2-sulfonyl)-3-nitro-1,2,4-triazole (MSNT) as a coupling reagent, and this was followed by treatment with aqueous ammonia [13].
Mono-6-azidocyclodextrins are accessible through a reaction of α, β, or γ-cyclodextrins with lithium azide, triphenylphosphine, and carbon tetrabromide [14]. However, di- and triazido cyclodextrins are formed as well, making a purification step necessary. Azido cyclodextrins can be readily reduced to the corresponding amino derivatives using the Staudinger reaction [15] or hydride donors [16] and are good 1,3-dipoles for click chemistry.
Together with tosylation, halogenation is the most common starting point for cyclodextrin functionalization. The most convenient and general route to these halo derivatives uses N-halosuccinimides as the halogenating reagent [17]. These halides (and also the tosylates) can be readily transformed by nucleophilic substitution. For instance, their reaction with an excess of sodium or lithium azide quantitatively provides an alternative route to mono-6-deoxy-6-azido-cyclodextrins.
Selective mono-6-esterification of β-cyclodextrin has been achieved through its reaction with maleic acid or itaconic acid to selectively yield the corresponding monoesters using phosphate as a catalyst in a semi-dry process [18].
In another approach, selective protection of the secondary rim makes the primary rim readily available for modifications. Taking advantage of their higher basicity, the secondary hydroxyl groups at position 2 of β-cyclodextrins can be selectively protected with t-butyldimethylsilyl chloride using NaH as base. The introduction of these bulky groups allows for the primary hydroxyls to undergo reactions with electrophiles without competition from the C-2 or C-3 hydroxyls. The mild removal of silyl protective groups from the secondary rim is then accomplished using tetrabutylammonium fluoride [19].
2.2. Disubstitution at C6-OH
Compared with monosubstitution, the synthesis of disubstituted cyclodextrins entails an additional issue: the formation of regioisomers, which need to be separated using chromatographic techniques. However, in some cases it is possible to guide the reaction toward the formation of a specific regioisomer.
Some of the first cyclodextrin capping strategies involved the introduction of aromatic residues from aromatic disulfonyl chloride reagents. Highly regioselective capping was found in the case of benzophenone-3,3′-disulfonylchloride, which mainly resulted in the AC regioisomer, while biphenyl-4,4′-sulfonyl chloride mainly afforded the AD capping product (Figure 3) [20].
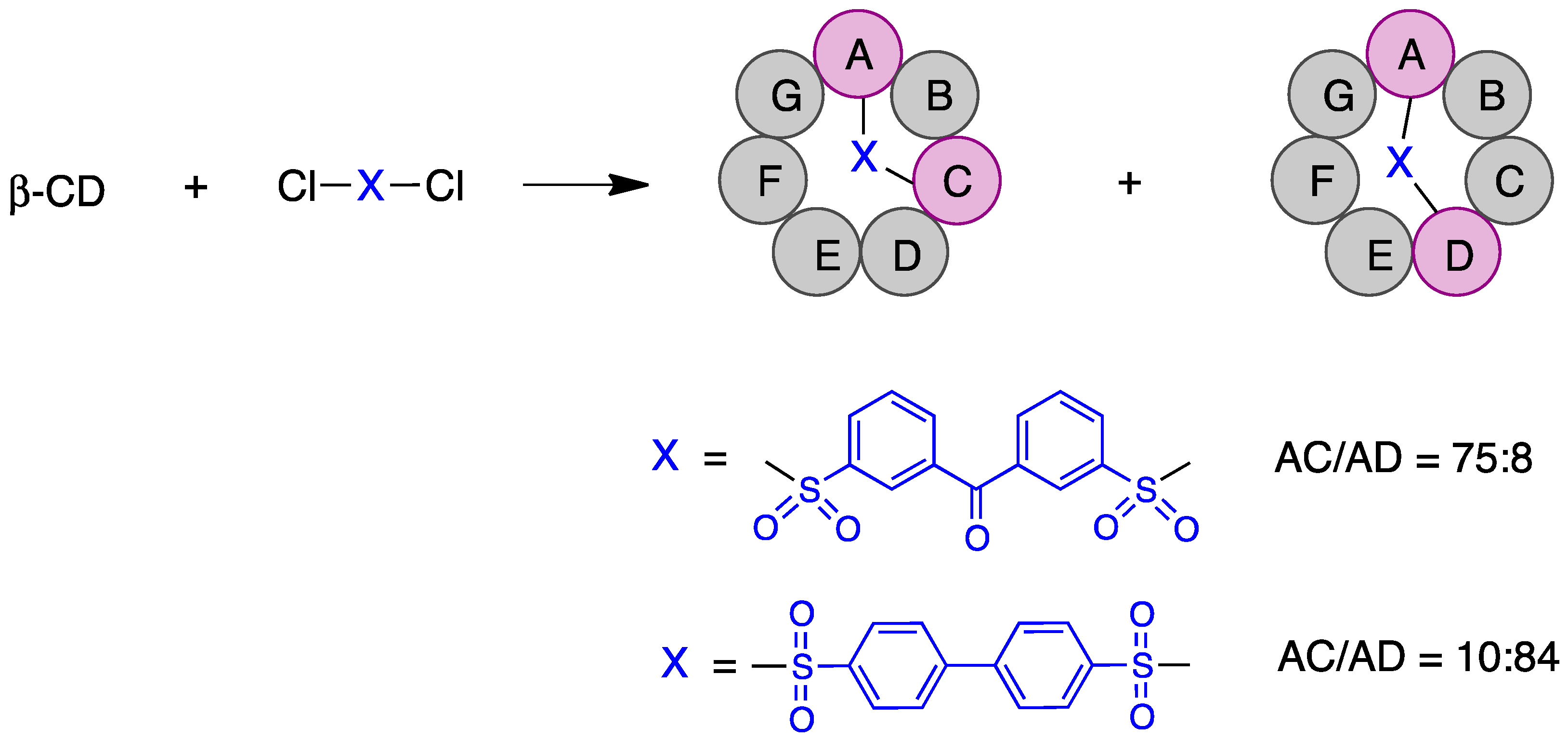
Figure 3. Regioselective AC and AD capping of β-cyclodextrin.
Diiodide derivatives obtained from disulfonyl cyclodextrins can also serve as substrates for further modifications through nucleophilic attack. Sulfonyl groups are displaced via reaction with iodide ion to yield the corresponding diiodo cyclodextrins [21]. Similarly, diazido cyclodextrins can be readily obtained from sulfonyl derivatives [22].
The selective synthesis of AD diols at the primary side is achieved through a DIBAL-H-mediated regioselective deprotection of the primary hydroxyl groups of perbenzylated α, β, and γ cyclodextrins [23]. For steric reasons, this bis-debenzylation takes place at the opposite glucose rings and involves two independent Lewis acid-assisted debenzylations (Figure 4). It was possible to stop the deprotection after the first debenzylation and isolate the monohydroxy cyclodextrin [24].
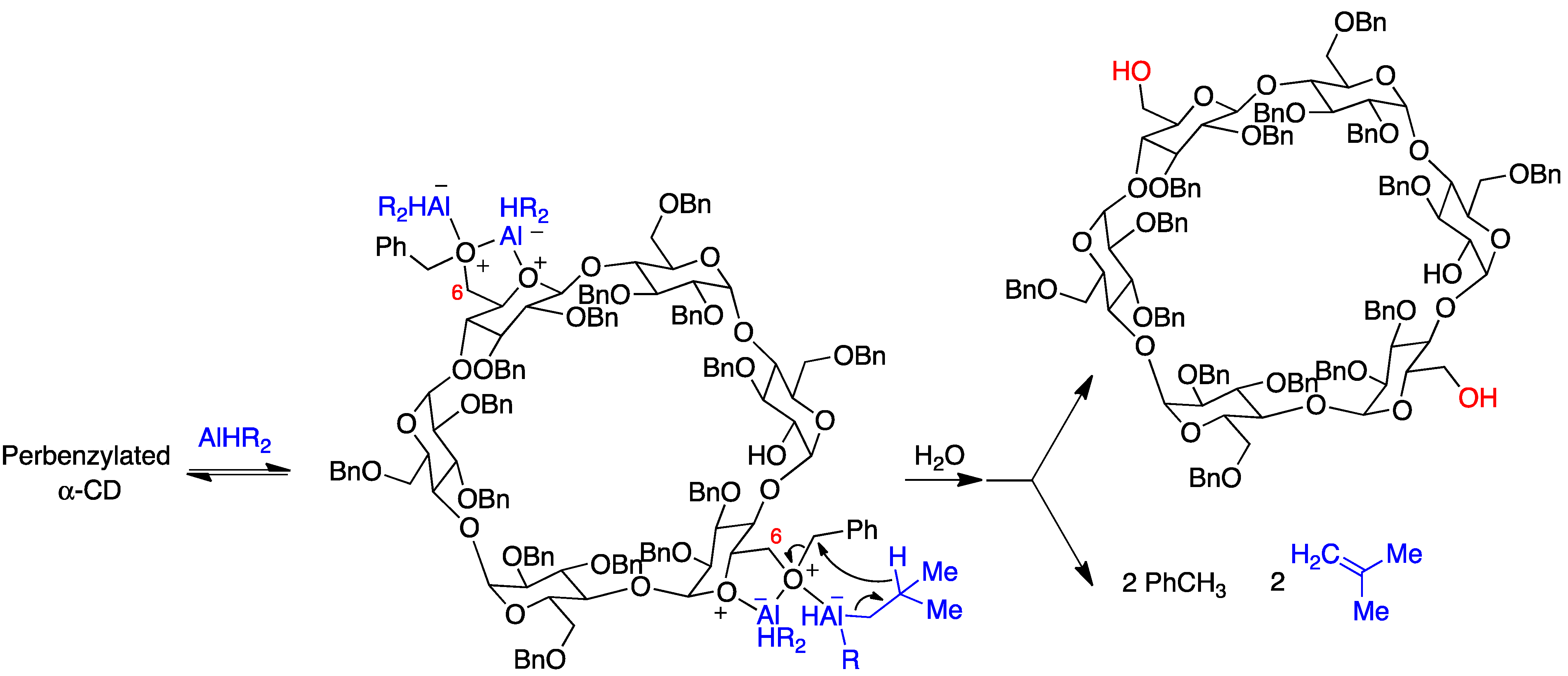
Figure 4. Selective debenzylation of the AD primary hydroxyls in α-cyclodextrin by DIBAL-H.
Perbenzylated α-cyclodextrins can be functionalized using an iterative process that predictably directs the debenzylation steps to access cyclodextrins with two, three, and five points of attachment for one, two, or three different new functions [25]. In a refinement of the method, through five consecutive site-directed debenzylations mediated by diisobutylaluminium hydride (DIBAL-H), a 2,3-tetradecabenzylated cyclodextrin hexa-hetero-functionalized with six different substituents at the primary hydroxyls was accessed [26].
2.3. Other C6-OH Substitution Patterns
Trisubstitution of cyclodextrins is a difficult goal for the variable substitution degree the reactions provide as well as for the generation of numerous positional isomers. However, some methods have successfully introduced three substituents in the primary side of cyclodextrins. Thus, complex di- and trifunctionalization patterns on the primary side of α-cyclodextrins are readily accessible owing to selective, iterative deprotections of perbenzylated α-cyclodextrin, where the first steps guide the selectivity of the subsequent ones. These modifications allow to selectively deprotect certain positions and also introduce deoxymethyl and deoxyvinyl residues at C-6 [26]. Similarly, perbenzylated α-cyclodextrins were selectively deprotected to yield a variety of primary-rim-functionalized compounds such as an ABD trihydroxy perbenzylated cyclodextrin and an AD diol, CF O-diTBS-protected perbenzylated cyclodextrin containing three pairs of functionalities on its primary rim [27].
Regarding the complete functionalization of primary alcohols at the primary rim of cyclodextrins, it has, in principle, the advantage that no positional isomers are generated. Nevertheless, factors such as steric crowding and complex formation decrease the yield as the degree of substitution increases. Sulfonylation with tosyl chloride is a good approach to perfunctionalize cyclodextrins, and has been applied to α- [28], β- [29], and γ- [30] cyclodextrins. Persulfonylated cyclodextrins are generally used as intermediates toward other functionalized CD derivatives.
Perhalogenated cyclodextrins also represent a good intermediate for further functionalization, with a higher stability than sulfonyl derivatives but with compromised solubility in non-polar solvents. Direct per-6-iodination of α and β-cyclodextrins can be achieved through reaction with iodine and triphenylphosphine in DMF. Similarly, the reaction of β-cyclodextrin with triphenylphosphine and bromine yields the corresponding per-6-brominated β-cyclodextrin [31]. α, β, and γ-cyclodextrins react with chloromethylenemorpholinium chloride and bromomethylenemorpholinium bromide in DMF to furnish the per-6-chlorinated and per-6-brominated cyclodextrins, respectively [32]. These per-6-halo cyclodextrins are very useful starting materials for additional functionalization processes such as the synthesis of per-amino derivatives and amino acid derivatives [17]. The use of per-6- iodo-per-6-deoxy-β- and γ-cyclodextrins for the synthesis of azido or mercapto derivatives has been performed under solvent-free conditions in a planetary ball mill. This mechanochemical protocol simplified the isolation and purification processes, while also allowing an efficient scale-up [33].
Per-6-alkylation of cyclodextrins is not a straightforward transformation and requires the adoption of a protection–deprotection strategy. An example with α-cyclodextrin involves, in this order, selective protection of the primary hydroxyls as silyl ethers, benzylation of the secondary hydroxyls, selective desilylation of the primary side, reaction of the primary hydroxyls with methyl iodide under strongly basic conditions (sodium hydride), and a final hydrogenolysis of the benzyl ethers [34].
The selective introduction of azido groups at all primary hydroxyls is achieved by the reaction of β-cyclodextrin with triphenylphosphine, lithium azide, and carbon tetrabromide in DMF with further treatment with acetic anhydride to yield heptakis(6-azido-6-deoxy)-β-cyclodextrin-tetradeca(2,3)acetate [35]. Per-azido cyclodextrins have been investigated as substrates for copper-catalyzed azide–alkyne cycloadditions [36].
3. Selective Modification of the Secondary Rim
In general, chemical modification of this side of cyclodextrins is not as straightforward as at the primary rim. This side is more crowded and hydroxyl groups at positions 2 and 3 establish hydrogen bonds making it more rigid. Furthermore, 2 and 3 hydroxyls differ in their reactivity: while the hydroxyl group C-2 is the most acidic of the glucopyranose and is selectively deprotonated with strong bases such as NaH, the C-3 hydroxyl is the least reactive.
3.1. Monosubstitution at C2-OH
β-cyclodextrin can be transformed into the corresponding 2-monotosyl-β-cyclodextrin via reaction with m-nitrophenyl tosylate [37]. The ability of cyclodextrins to form complexes with chemical reagents through the hydrophobic cavity modifies their reactivity, and this feature is used here to selectively direct the reaction to the secondary rim of the cyclodextrin instead of the more reactive primary rim. This complex-formation-based approach was optimized with the design of a more efficient tosyl donor, 1-(p-tolylsulfonyl)-1,2,4-triazole [38]. An additional method to selectively synthesize monotosyl α and β-cyclodextrins involves their reaction with N-tosylimidazol in DMF and molecular sieves [39] or under solid-state conditions, using mechanochemical activation [40]. 2-Monotosyl cyclodextrins are key intermediates for the functionalization of not only C-2 but also the C-3 position through the formation of an epoxide.
Direct alkylation at position 2 of β-cyclodextrin is achieved through its reaction with dialkyl sulfates in dilute aqueous alkali [41]. An indirect method to reach position 2, as well as position 3, is the regioselective bis-O-demethylation of 2,3,6-permethylated α and β-cyclodextrins with DIBAL-H in toluene to yield 2A,3B-diol permethylated cyclodextrins as major products, allowing for selective functionalization of a single 2 hydroxyl and/or a single 3 hydroxyl [42][43].
3.2. Disubstitution at C2-OH
The reaction in DMF of γ-cyclodextrin with benzophenone-3,3-disulfonylimidazole, a rigid disulfonyl precursor, selectively yields 2AB-disulfonylated γ-cyclodextrin [44]. A related rigid disulfonyl imidazole reagent, 1,4-dibenzoyl benzene-3′,3″-disulfonyl imidazole, selectively orients the sulfonylation toward the positional isomer 2AC in α, β, and γ-cyclodextrins [45]. The design of a longer disulfonyl imidazole bridge yielded the positional isomer 2AD in α, β, and γ-cyclodextrins, with a better selectivity for β [46]. These 2-disulfonyl cyclodextrins can serve as versatile building blocks for further modifications at position 2 as well as position 3 through the formation of an intermediate epoxide or by treatment with ammonia to yield the corresponding amine.
3.3. Persubstitution at C2-OH
Persubstitution at position 2 is favored by the use of strong bases that deprotonate C2-hydroxyls. However, the successive introduction of electrophiles makes the secondary rim increasingly crowded, making subsequent substitutions more difficult and shifting the reactivity toward the primary hydroxyls. To avoid this, per-2-tosylations and per-2-alkylations are often carried out after the protection of primary hydroxyls with silyl groups, which can be deprotected afterward [47]. An exception is the selective per-2-methylation of β-cyclodextrin with methyl iodide, sodium hydride, and DMSO [48], or using N-tosylimidazole and a catalytic amount of Cs2CO3 in DMF [49].
A different approach for the selective per-2-alkylation of cyclodextrins starts with the selective per-2,6-silylation of β-cyclodextrin with tert-butyldimethylsilyl chloride and pyridine. Then, during the reaction with methyl iodide and sodium hydride, the silyl groups at the C-2 hydroxyls migrate to the C3-OH groups, and the deprotected position 2 is selectively methylated. A final desilylation affords the per-2-methylated cyclodextrin [50]. This C2-OH to C3-OH migration is also observed in other cases where a more complex substitution pattern is present [51][52].
3.4. Monosubstitution at C3-OH
C-3 in cyclodextrins are the least reactive hydroxyls in the glucopyranose ring due to their low accessibility. A common strategy to functionalize position 3 consists of the tosylation at C2-OH and treatment with a base to generate an epoxide, followed by the introduction of nucleophiles [53]. This provides a mixture of 2- and 3-substituted products that can be separated with chromatographic techniques, with substitution at position 3 being highly favored in most cases. The combination of N-benzoylimidazole and carbonate buffer in DMF gave mainly acylation C-3 hydroxyl groups of β-CD, which was ascribed to acylation at both C2-OH and C3-OH accompanied by C2-OH to C3-OH acyl migration [54]. In other transformations, reagent complexation in the cyclodextrin cavity may direct the reaction to C3-OH. For instance, the 3-sulfonylation of α-cyclodextrins is achieved by direct reaction with β-naphtalenesulfonyl chloride [55]. A particular case of alkylation is the selective introduction of an alkyl group at just one 3 position of β-cyclodextrins through the reaction of per-6-terbutyldimethylsilylated β-cyclodextrin with N-methyl-4-chloromethyl-2-nitroaniline in the presence of lutidine [56].
3.5. Di- and Trisubstitution at C3-OH
In a similar way to that of monosubstitutions at position 3, the manno-2,3-epoxycyclodextrin is a common intermediate used for the introduction of nucleophiles. The positional isomers obtained depend on the initial 2-ditosylate involved in the route. Also, 3-sulfonylation of α-cyclodextrins is achieved via reaction with β-naphtalenesulfonyl chloride and further isolation of the 3-disulfonylated derivatives, particularly a mixture of positional isomers 3AC and 3AD disulfonyls that are obtained due to an oriented synthesis owing to complex formation between the β-cyclodextrin and sulfonyl chloride [57]. The mixture of regioisomers is easily resolved with reverse-phase column chromatography.
The sulfonylation of β-cyclodextrin with β-naphtalenelsulfonyl chloride in basic aqueous acetonitrile was selectively found to yield the 3ACE-trisulfonylated isomer [58].
3.6. Persubstitution at C3-OH
Perfunctionalization at position 3 of cyclodextrins is a challenging task because 3-hydroxyls are the least reactive and are involved in hydrogen bonding with 2-hydroxyls. Furthermore, they are located in a crowded side of the cyclodextrin which becomes even more crowded after protection of 2-hydroxyls, hindering further reactions.
Despite all these setbacks, perbenzylated α-cyclodextrin can be selectively deprotected at position 3 with Et3SiH and I2 to provide a 2A–F,6A–F-dodeca-O-benzyl α-cyclodextrin. This strategy works as well starting from a 2A–F,3A-F,6A-E-heptadec-O-benzyl α-cyclodextrin and a 2A–F,3A-F,6BCEF-hexadec-O-benzyl α-cyclodextrin to yield the corresponding 3-deprotected derivatives with one or two deprotected hydroxyls in the primary side, respectively. These derivatives stand out as useful starting materials for the selective perfunctionalization of the relatively unreactive position 3 [59].
4. Persubstitution of the Primary and Secondary Rims
Although steric hindrance of persubstituted cyclodextrins make the introduction of substituents increasingly difficult, the absence of regioisomers is a major advantage in the synthesis of 2,3,6-perfunctionalized cyclodextrins. Perbenzylation of α and β-cyclodextrins is performed in excellent yield (>90%) by treatment with benzyl chloride and sodium hydride [25]. Similarly, peralkylation of β-cyclodextrin is achieved in 74% yield via reaction with methyl iodide and sodium hydride in DMSO [60].
5. Cyclodextrin-Based Polymers
CD-based polymers are the basis for new CD-based materials for different types of application fields for pharmaceutical and biomedical applications, but are also useful in cosmetic and food science [61][62][63].
5.1. Radical Polymerization Reactions
Atom transfer radical polymerization (ATRP) is a type of radical polymerization having as the key step a transition metal-catalyzed C-C bond generation. Generally, an alkyl halide R-X is used as the initiator and a complex formed by a transition metal (e.g., CuI) and a chelating agent is used to generate radicals via a one-electron transfer process, while simultaneously the transition metal is oxidized to a higher oxidation state (Figure 5).
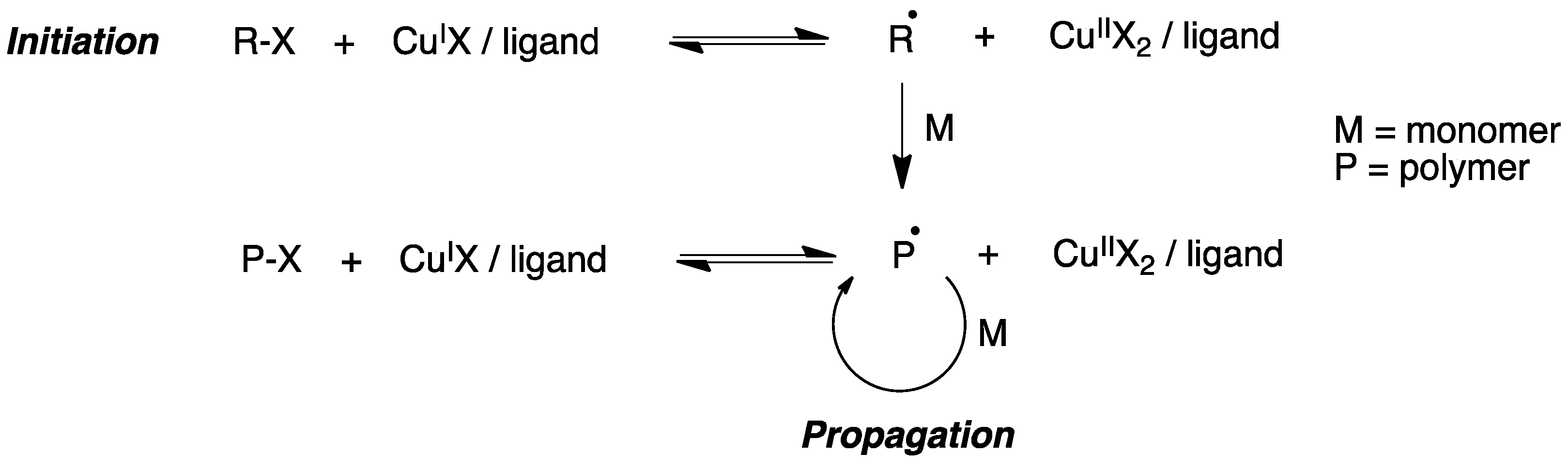
Figure 5. Mechanism of atom transfer radical polymerization (ATRP).
α-Haloester moieties attached to cyclodextrin-based monomers have been widely employed for performing ATRP processes. These moieties can be introduced by direct per-acylation of β-CD with 2-bromoisobutyryl anhydride (Figure 6A) [64]. Alternatively, the primary rim can be selectively functionalized starting from the corresponding mercaptocyclodextrin using a radical thiol–ene reaction (Figure 6B) [65]. As an example of one of these polymerizations, the researchers summarize in Figure 6C the synthesis of a 21-arm star polymer via a core-based approach, with the core being a molecule of β-cyclodextrin. To this end, heptakis [2,3,6-tri-O-(2-bromo-2-methylpropionyl]-β-cyclodextrin, prepared via the peracylation method described above, was treated with methyl methacrylate in the presence of copper bromide and n-propyl-2-pyridylmethanimine as a bidentate ligand for copper complexation.
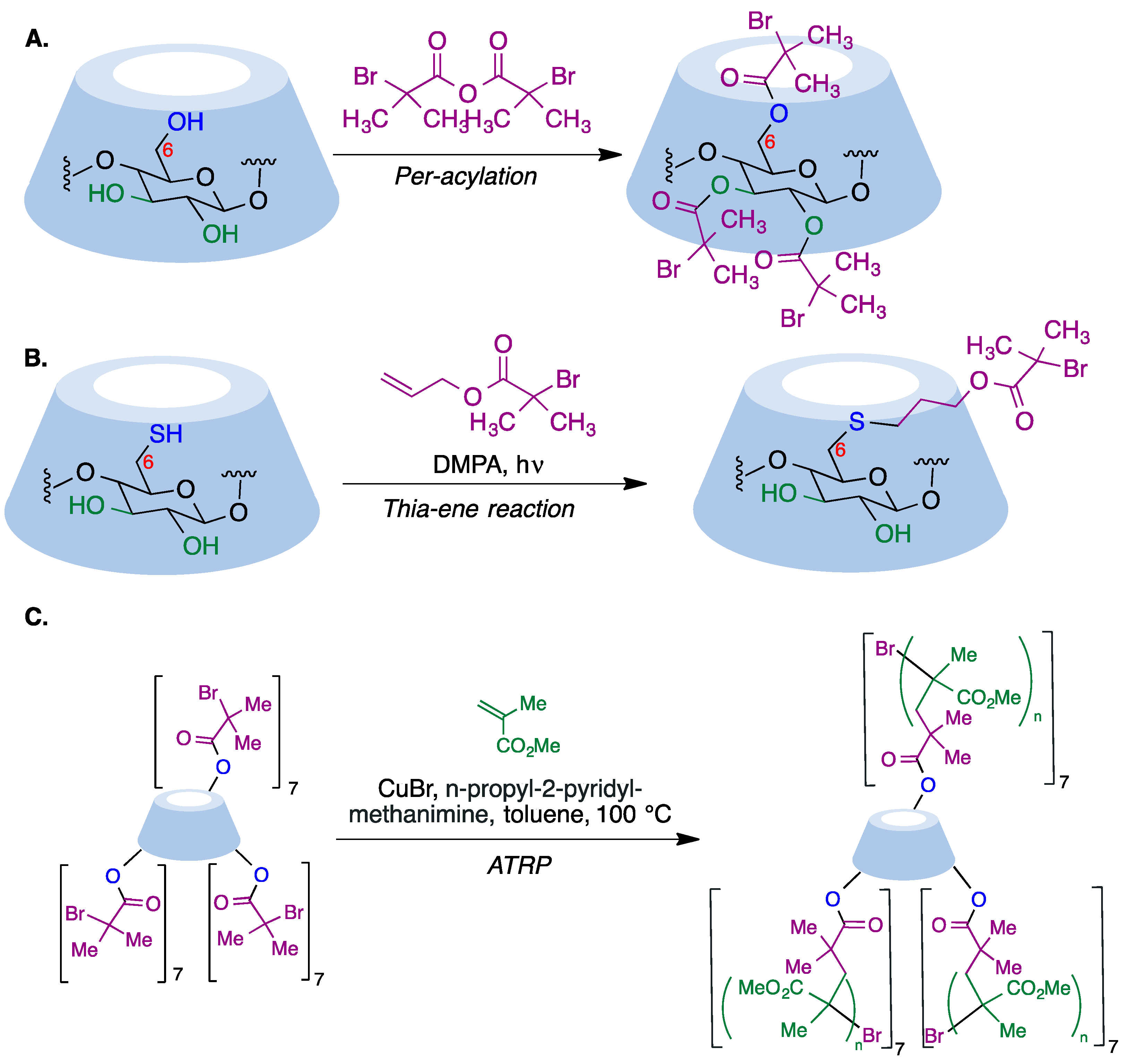
Figure 6. (A) Synthesis of per-acylated β-CD via treatment with 2-bromoisobutyryl anhydride. (B) Selective functionalization of the cyclodextrin primary rim using a radical thiol–ene reaction. (C) Core-based synthesis of a 21-arm star polymer using the ATRP method.
In a reverse approach, the acrylate moiety required for the polymerization process can be introduced first into the cyclodextrin. Thus, treatment of 6-monopiperazino-β-cyclodextrin, obtained from the corresponding 6-monotosyl derivative, with glycidyl methacrylate afforded a mono-methacrylate substituted cyclodextrin via epoxide opening (Figure 7A) [66]. A similar type of starting material can be obtained by means of copper-mediated azide–alkyne couplings (CuAAC) of azido-cyclodextrins (Figure 7B) [67].
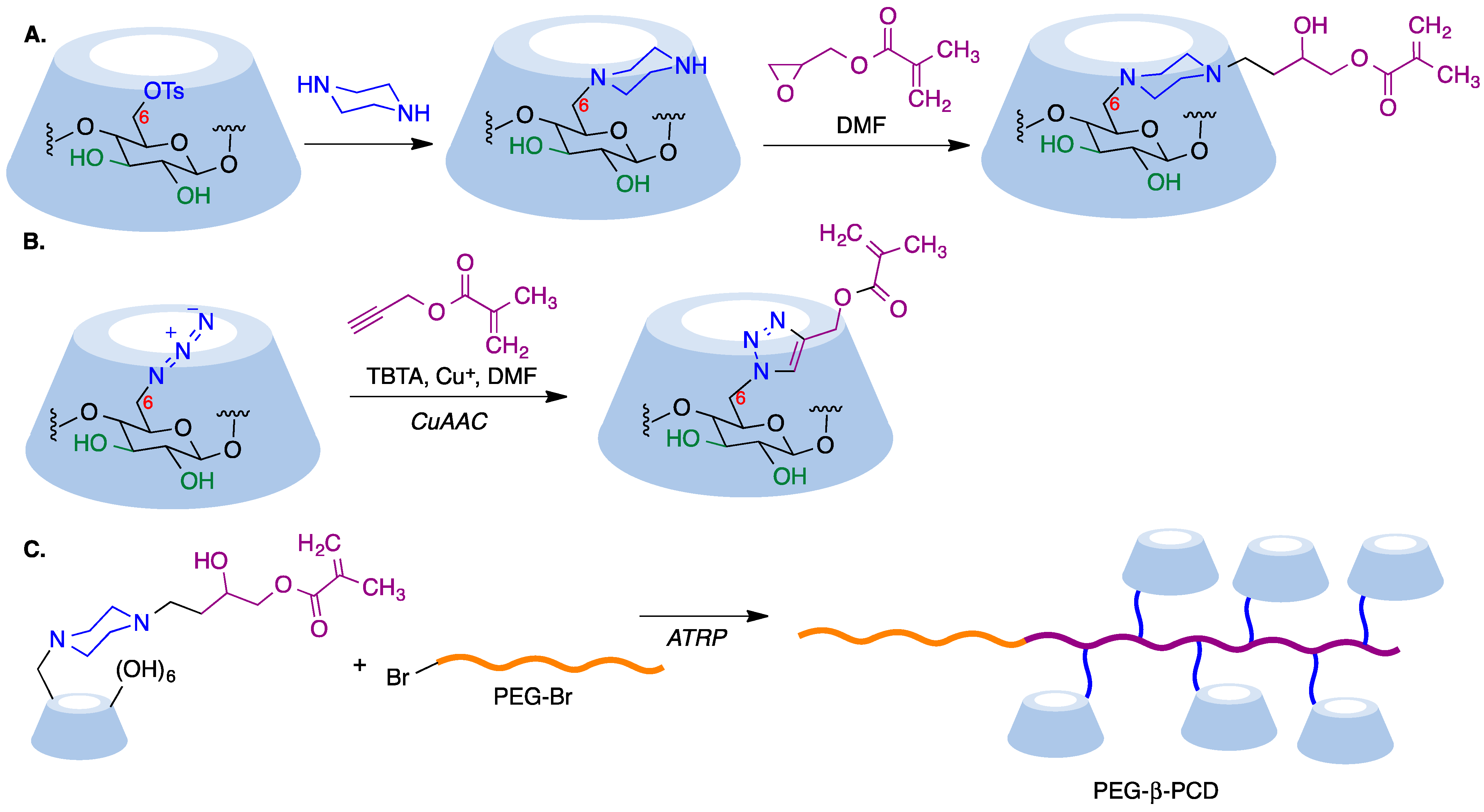
Figure 7. (A) Synthesis of a β-cyclodextrin derivative containing an acrylate moiety by opening of the epoxide moiety in glycidyl methacrylate with 6-monopiperazino-β-cyclodextrin. (B) Synthesis of a β-cyclodextrin derivative containing an acrylate moiety via a CuAAC reaction. (C) Synthesis of a CD-pendant polymer using the ATRP method.
Mono-methacrylate cyclodextrins are useful as starting materials for radical polymerization reactions. Thus, poly(ethyleneglycol)-β-poly(cyclodextrin) (PEG-β-PCD), can be easily synthesized via atom transfer radical polymerization using a poly(ethylene glycol) macroinitiator (Figure 7C). This well-defined, hydrophilic, CD-pendant copolymer was able to include a variety of guest molecules and self-assembled into advanced nanostructures due to the synergistic effect of CD moieties [67].
Reversible addition–fragmentation chain transfer (RAFT) polymerization is another relevant modality of radical-induced polymerization that has proved to be an excellent tool to access a range of well-defined macromolecular architectures. This technique involves the use of a chain-transfer agent, usually a thiocarbonylthio compound, which helps to achieve control over the generated molecular weight and polydispersity during free-radical polymerization (Figure 8) [68].
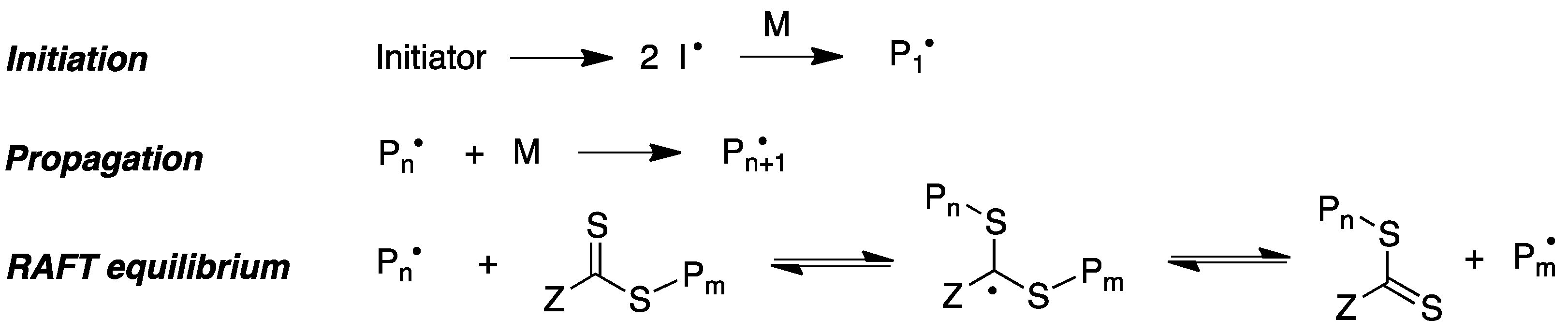
Figure 8. Simplified mechanism of the RAFT polymerization process.
Thiocarbonylthio functional groups can be installed onto cyclodextrin molecules by a simple acylation process using benzyl (3-chloro-3-oxopropyl) carbonotrithioate, an acid chloride [69], or the corresponding acid in the presence of dicyclohexylcarbodiimide (DCC) and hydroxybenzotriazole (HOBT) as a coupling reagent [70]. RAFT polymerization of this starting material in the presence of 2-hydroxyethyl acrylate (HEA), using 2,2′-azobisiobutyronitrile (AIBN) as initiator, afforded CD-based polymers with seven-arm star architectures [70] (Figure 9).
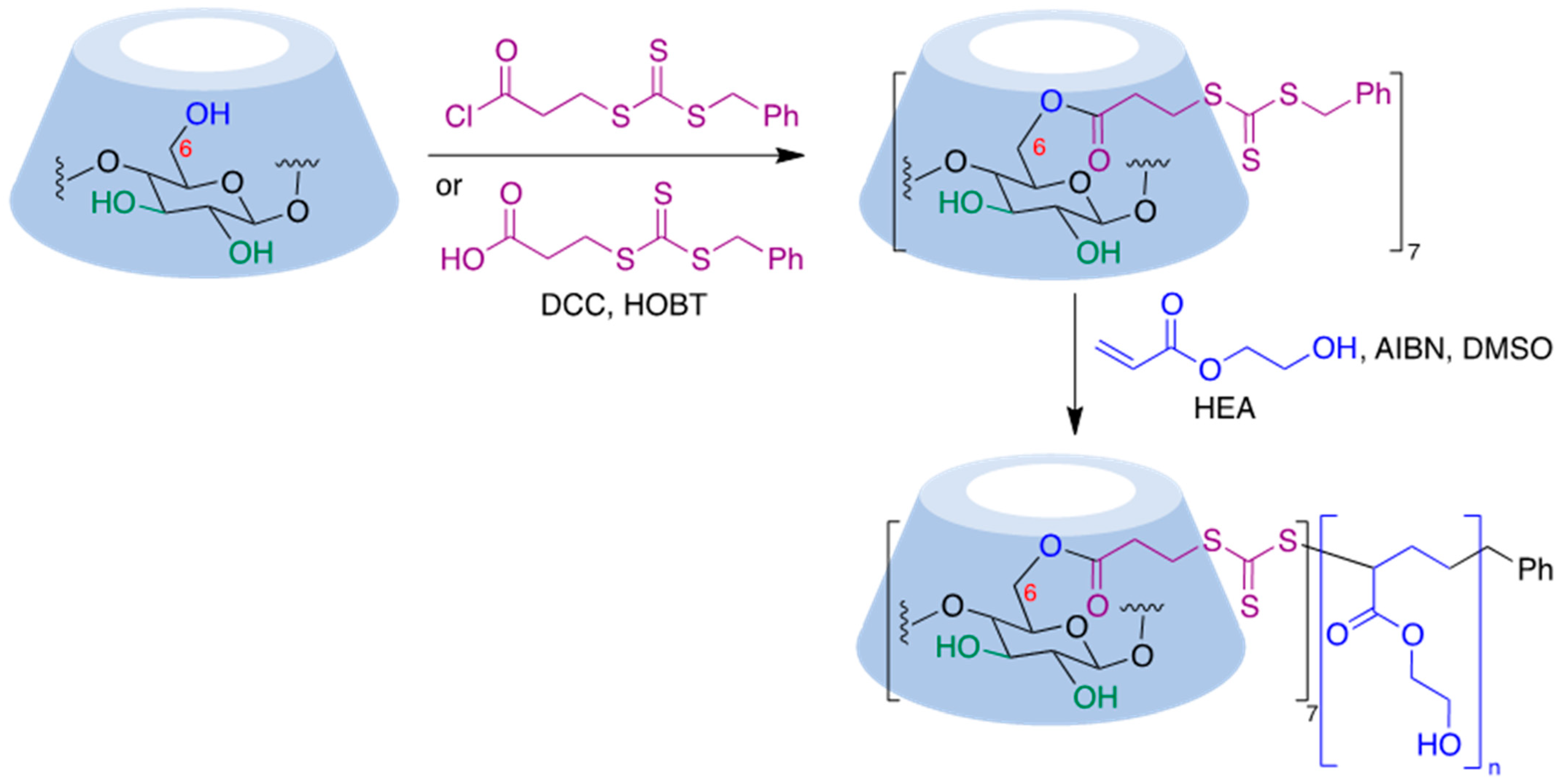
Figure 9. Application of the RAFT polymerization process to a cyclodextrin substrate.
5.2. Ring-Opening Polymerization (ROP)
Cyclodextrins can serve as core structures for anionic ring-opening polymerization (ROP) processes, which involve CD derivatization using cyclic esters [71]. Thus, treatment of β-cyclodextrin acetylated at its secondary rim with ε-caprolactone in the presence of Sn(Oct)2 as a catalyst resulted in polymers with narrow dispersity by esterification of the primary rim. The polymer arms were then coupled with DCC to carboxyl-terminated poly(ethylene glycol) to form amphiphilic copolymers that were able to undergo micellization (Figure 10) [72]. The ring opening polymerization of ε-caprolactone can also be carried out in the presence of wet β-cyclodextrin and has been performed in pressurized (12–13 MPa) batch reactors [73]. Lactide (3,6-dimethyl-1,4-dioxane-2,5-dione) is also widely employed to generate cyclodextrin-based polymers and, in the presence of 4-dimethylaminopyridine as an organocatalyst, has allowed the quantitative functionalization of α-, β-, γ-, and 2,3-dimethyl-β-cyclodextrins, with narrow molecular weight distributions (dispersity < 1.1) [74].
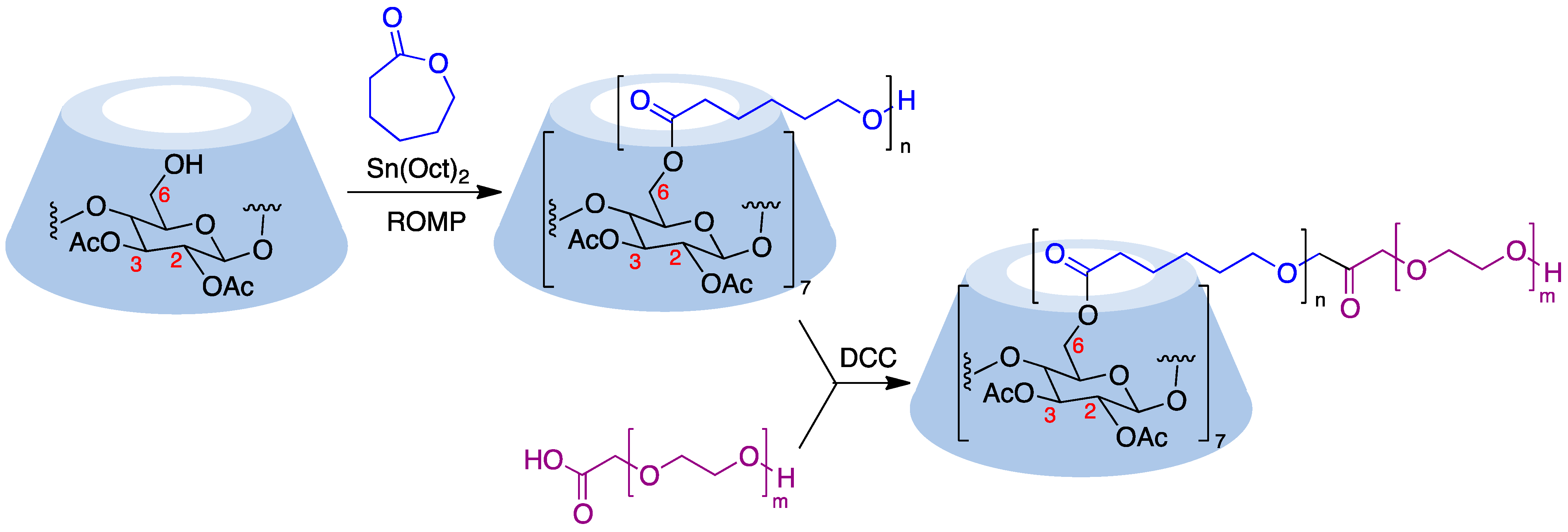
Figure 10. Ring-opening polymerization carried out on a cyclodextrin substrate.
5.3. CuAAC-Based Polymerization
Although ATRP is a suitable tool for the core-first synthesis of multiarmed polymers, it has some limitations because the polymerization of many arms from one core can lead to steric hindrance and a high local concentration of free radicals that facilitates star–star coupling and other termination events. This can be improved by use of the so-called grafting-to approach, where a functional core is coupled to a functional polymer, where the most extended reaction for such coupling is the copper-catalyzed azide–alkyne cycloaddition (CuAAC) [75], which is frequently associated with ATRP polymerization. To this end, a polymer containing an alkyne moiety joined to poly(N-isopropylacrylamide) (PNIPAM) is prepared via the ATRP polymerization technique (Figure 11A) and this precursor is then coupled to an azido-CD, which can be prepared via nucleophilic displacement of halogens at the primary rim of cyclodextrins (Figure 11B). In order to involve all cyclodextrin hydroxy groups, the primary rim can be modified via the CuAAC reaction and the secondary rim is used as the substrate of an ATRP or ROP polymerization. Alternatively, per-acylation with 2-bromopropionic bromide followed by bromide nucleophilic displacement with sodium azide affords a per-azidocyclodextrin, which is then submitted to a CuAAC reaction with the alkyne-PNIPAM polymer to furnish a 21-star polymer (Figure 11C) [76].
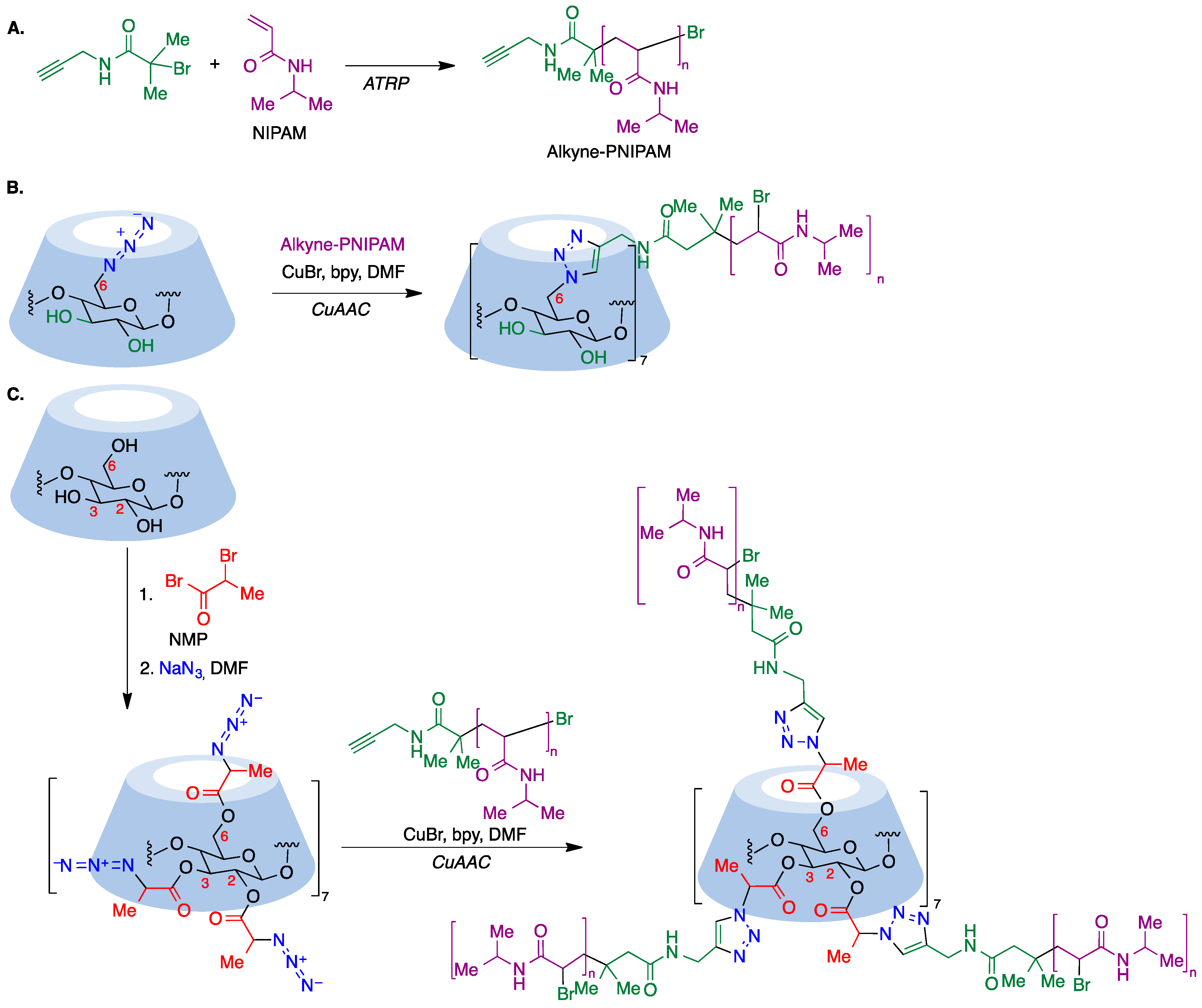
Figure 11. (A) Synthesis of PNIPAM, a polymer containing an alkyne moiety joined to poly(N-isopropylacrylamide. (B) Coupling of PNIPAM to a primary rim azido-CD via the CuAAC reaction. (C) Synthesis of a 21-star polymer via a CuAAC reaction of a per-azidocyclodextrin with PNIPAM.
6. Cyclodextrin-Embedded Covalently Crosslinked Networks
Hydrogels are three-dimensional crosslinked polymeric networks [77][78]. These biomaterials show unique characteristics such as their biocompatibility, water retention ability, and morphological similarity to bodily tissues that make them very useful for many biomedical applications. The incorporation of cyclodextrins, combining hydrophilic exteriors with hydrophobic inner pockets able to form inclusion complexes, provides an interesting approach to the design of hydrogels. The main crosslinking methods employed to prepare CD-based materials are summarized in Figure 12 and include radical polymerization, CuAAC-based polymerization, and polymerization processes based on nucleophilic addition or substitution reactions.
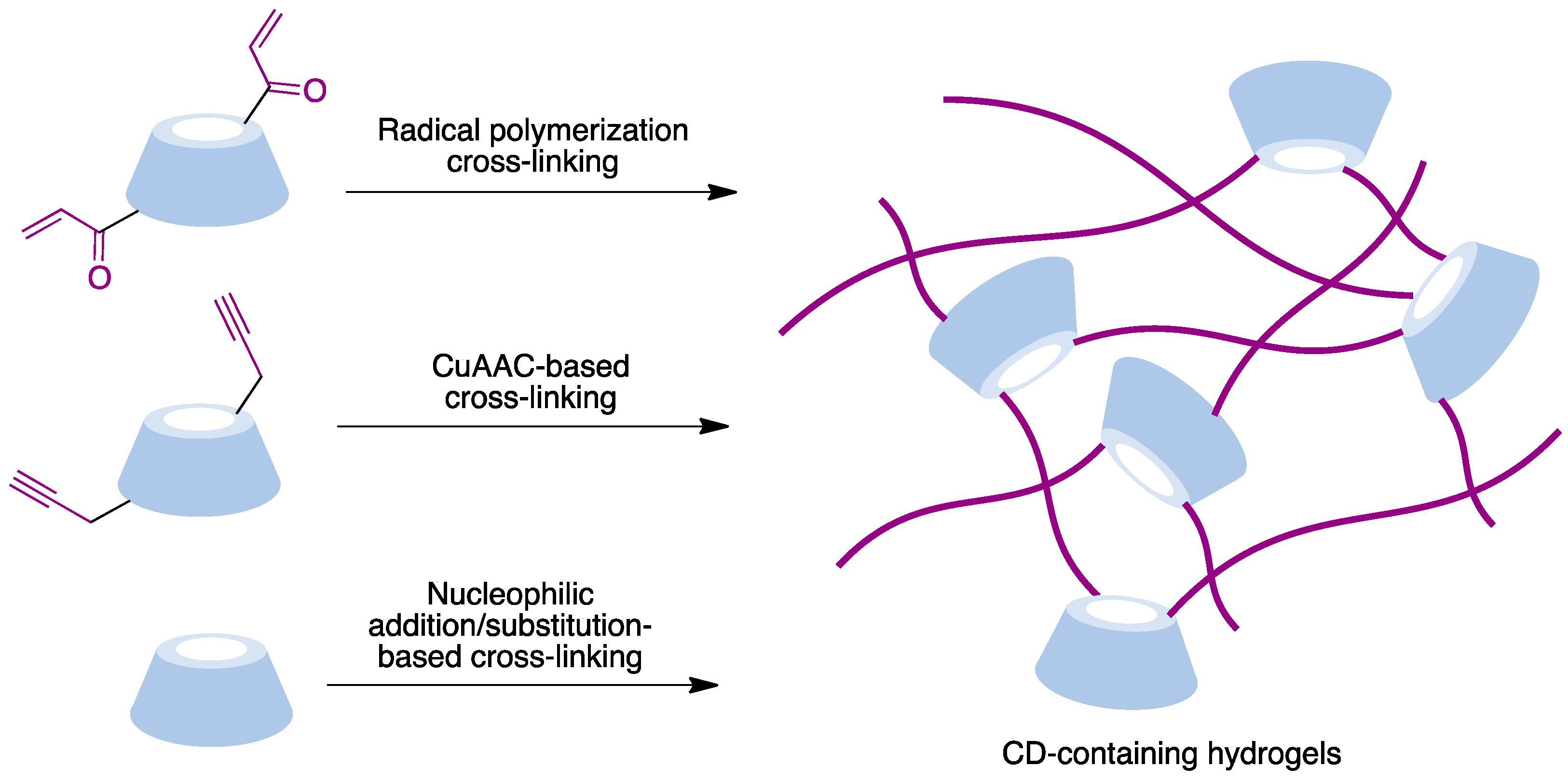
Figure 12. Main methodologies to access covalently CD-embedded hydrogels.
One interesting example of cyclodextrin-based networks are nanosponges, which can be described as colloidal structures that contain solid, three-dimensional, hyper-reticulated nanoporous structures that protect the entrapped molecules from degradation and help to improve the solubility of lipophilic compounds [79]. Cyclodextrin-based nanosponges (CDNSs), in particular, can be efficiently engineered for drug delivery purposes, especially in cancer therapy [80][81][82]. Indeed, due to the existence of lipophilic cavities in the cyclodextrin building blocks and the possibility to install a hydrophilic network in the nanosponge itself, depending on the nature of the crosslinker, these materials are ideal candidates to increase the stability of sensitive and volatile compounds, as well as the solubility of both lipophilic and lipophobic compounds [83][84].
The synthesis of these nanostructures is achieved following the general scheme shown in Figure 13, where a suitable difunctional cross-linking reagent generates covalent bridges between cyclodextrin molecules by reacting with their hydroxyl groups to generate carbamate, carbonate, diester, or diether links. These transformations can be promoted under a number of experimental conditions, including, among others, solution conditions in conventional or non-conventional solvents, the joint melting of the CD and the linker [85], and ultrasound-assisted [86], microwave-assisted [87], and mechanochemical [88] conditions.
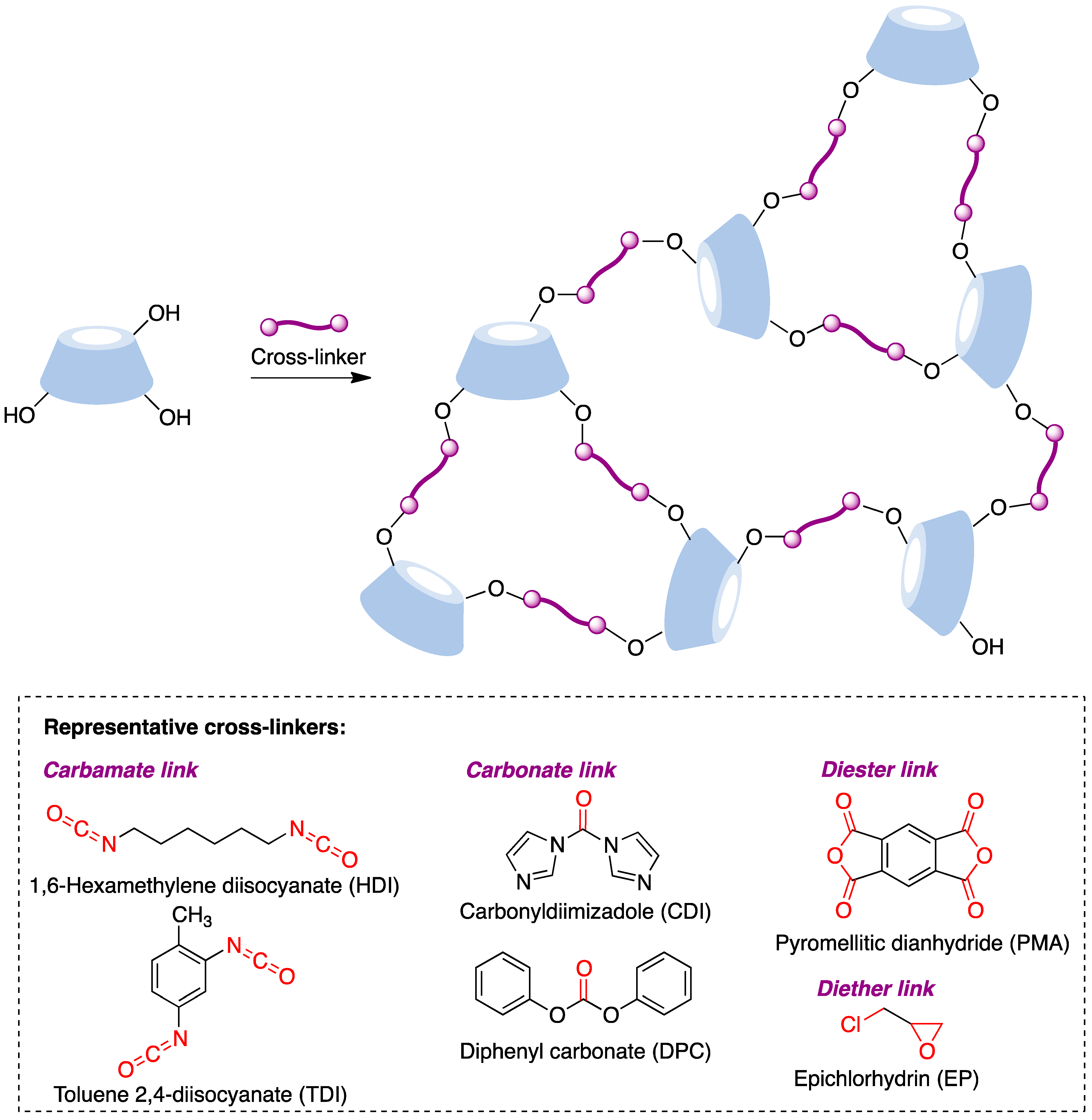
Figure 13. Structure and synthesis of CD-based nanosponges.
This entry is adapted from the peer-reviewed paper 10.3390/pharmaceutics15092345
References
- Xu, W.; Li, X.; Wang, L.; Li, S.; Chu, S.; Wang, J.; Li, Y.; Hou, J.; Luo, Q.; Liu, J. Design of cyclodextrin-based functional systems for biomedical applications. Front. Chem. 2021, 9, 635507.
- Khan, A.R.; Forgo, P.; Stine, K.J.; D’Souza, V.T. Methods for selective modifications of cyclodextrins. Chem. Rev. 1998, 98, 1977–1996.
- Liu, J.Y.; Zhang, X.; Tian, B.R. Selective modifications at the different positions of cyclodextrins: A review of strategies. Turk. J. Chem. 2020, 44, 261–278.
- Řezanka, M. Synthesis of substituted cyclodextrins. Environ. Chem. Lett. 2019, 17, 49–63.
- Wüpper, S.; Lüersen, K.; Rimbach, G. Cyclodextrins, natural compounds, and plant bioactives—A nutritional perspective. Biomolecules 2021, 11, 401.
- Martin, K.A.; Czarnik, A.W. Facile preparation of the β-cyclodextrinyl aldehyde. Tetrahedron Lett. 1994, 35, 6781–6782.
- Tripodo, G.; Wischke, C.; Neffe, A.T.; Lendlein, A. Efficient synthesis of pure monotosylated beta-cyclodextrin and its dimers. Carbohydr. Res. 2013, 381, 59–63.
- Nielsen, T.T.; Wintgens, V.; Amiel, C.; Wimmer, R.; Larsen, K.L. Facile synthesis of β-cyclodextrin-dextran polymers by “click” chemistry. Biomacromolecules 2010, 11, 1710–1715.
- Zhong, N.; Byun, H.S.; Bittman, R. An improved synthesis of 6-O-monotosyl-6-deoxy-β-cyclodextrin. Tetrahedron Lett. 1998, 39, 2919–2920.
- Cornwell, M.J.; Huff, J.B.; Bieniarz, C. A one-step synthesis of cyclodextrin monoaldehydes. Tetrahedron Lett. 1995, 36, 8371–8374.
- Yoon, J.; Hong, S.; Martin, K.A.; Czarnik, A.W. A general method for the synthesis of cyclodextrinyl aldehydes and carboxylic acids. J. Org. Chem. 1995, 60, 2792–2795.
- Sallas, F.; Leroy, P.; Marsura, A.; Nicolas, A. First selective synthesis of thio-β-cyclodextrin derivatives by a direct mitsunobu reaction on free β-cyclodextrin. Tetrahedron Lett. 1994, 35, 6079–6082.
- Di Fabio, G.; Malgieri, G.; Isernia, C.; D’Onofrio, J.; Gaglione, M.; Messere, A.; Zarrelli, A.; De Napoli, L. A novel synthetic strategy for monosubstituted cyclodextrin derivatives. Chem. Commun. 2012, 48, 3875–3877.
- Hanessian, S.; Benalil, A.; Laferriere, C. The synthesis of functionalized cyclodextrins as scaffolds and templates for molecular diversity, catalysis, and inclusion phenomena. J. Org. Chem. 1995, 60, 4786–4797.
- Guieu, S.; Sollogoub, M. Regiospecific tandem azide-reduction/ deprotection to afford versatile amino alcohol-functionalized α- and β-cyclodextrins. Angew. Chem. Int. Ed. 2008, 47, 7060–7063.
- Ashton, P.R.; Königer, R.; Fraser Stoddart, J.; Alker, D.; Harding, V.D. Amino acid derivatives of β-cyclodextrin. J. Org. Chem. 1996, 61, 903–908.
- Chmurski, K.; Defaye, J. An improved synthesis of per(6-deoxyhalo)cyclodextrins using N-halosuccinimides—Triphenylphosphine in dimethylformamide. Supramol. Chem. 2000, 12, 221–224.
- Li, Y.-F.; Ha, Y.-M.; Guo, Q.; Li, Q.-P. Synthesis of two β-cyclodextrin derivatives containing a vinyl group. Carbohydr. Res. 2015, 404, 55–62.
- Tian, S.; D’Souza, V.T. Selective protection of the secondary side of β-cyclodextrin. Tetrahedron Lett. 1994, 35, 9339–9342.
- Tabushi, I.; Yamamura, K.; Nabeshima, T. Characterization of regiospecific A,C- and A,D-disulfonate capping of β-cyclodextrin. Capping as an efficient production technique. J. Am. Chem. Soc. 1984, 106, 5267–5270.
- Breslow, R.; Canary, J.W.; Varney, M.; Waddell, S.T.; Yang, D. Artificial transaminases linking pyridoxamine to binding cavities: Controlling the geometry. J. Am. Chem. Soc. 1990, 112, 5212–5219.
- Benkovics, G.; Bálint, M.; Fenyvesi, É.; Varga, E.; Béni, S.; Yannakopoulou, K.; Malanga, M. Homo- and hetero-difunctionalized β-cyclodextrins: Short direct synthesis in gram scale and analysis of regiochemistry. Beilstein J. Org. Chem. 2019, 15, 710–720.
- Pearce, A.J.; Sinaÿ, P. Diisobutylaluminum-promoted regioselective de-O-benzylation of perbenzylated cyclodextrins: A powerful new strategy for the preparation of selectively modified cyclodextrins. Angew. Chem. Int. Ed. 2000, 39, 3610–3612.
- Lecourt, T.; Herault, A.; Pearce, A.J.; Sollogoub, M.; Sinaÿ, P. Triisobutylaluminium and diisobutylaluminium hydride as molecular scalpels: The regioselective stripping of perbenzylated sugars and cyclodextrins. Chem. Eur. J. 2004, 10, 2960–2971.
- Guieu, S.; Sollogoub, M. Multiple homo- and hetero-functionalizations of α-cyclodextrin through oriented deprotections. J. Org. Chem. 2008, 73, 2819–2828.
- Wang, B.; Zaborova, E.; Guieu, S.; Petrillo, M.; Guitet, M.; Blériot, Y.; Ménand, M.; Zhang, Y.; Sollogoub, M. Site-selective hexa-hetero-functionalization of α-cyclodextrin an archetypical C6-symmetric concave cycle. Nat. Commun. 2014, 5, 5354.
- Bistri, O.; Sinaÿ, P.; Sollogoub, M. Expeditious selective synthesis of primary rim tri-differentiated α-cyclodextrin. Tetrahedron Lett. 2006, 47, 4137–4139.
- Fujita, K.; Ohta, K.; Masunari, K.; Obe, K.-I.; Yamamura, H. Selective preparation of hexakis (6-O-arenesulfonyl) -α-cyclodextrin. Tetrahedron Lett. 1992, 33, 5519–5520.
- Yamamura, H.; Fujita, K. Preparation of heptakis(6-O-(p-tosyl))-β-cyclodextrin and heptakis(6-O-(p-tosyl))-2-O-(p-tosyl)-β-cyclodextrin and their conversion to heptakis(3, 6-anhydro)-β-cyclodextrin. Chem. Pharm. Bull. 1991, 39, 2505–2508.
- Yamamura, H.; Yoshitaka, K.; Masao, K.; Yasuo, B. Preparation of polytosylated γ-cyclodextrins. Bull. Chem. Soc. Jpn. 1993, 66, 585–588.
- Gadelle, A.; Defaye, J. Selective halogenation at primary positions of cyclomaltooligosaccharides and a synthesis of per-3,6-anhydro cyclomaltooligosaccharides. Angew. Chem. Int. Ed. Engl. 1991, 30, 78–80.
- Chmurski, K.; Defaye, J. An improved synthesis of 6-deoxyhalo cyclodextrins via halomethylenemorpholinium halides Vilsmeier-Haack type reagents. Tetrahedron Lett. 1997, 38, 7365–7368.
- Jicsinszky, L.; Caporaso, M.; Martina, K.; Gaudino, E.C.; Cravotto, G. Efficient mechanochemical synthesis of regioselective persubstituted cyclodextrins. Beilstein J. Org. Chem. 2016, 12, 2364–2371.
- Takeo, K.-I.; Ueraura, K.; Mitoh, H. Derivatives Of α-Cyclodextrin and the Synthesis of 6-O-α-D-glucopyranosyl-α-cyclodextrin. J. Carbohydr. Chem. 1988, 7, 293–308.
- Boger, J.; Corcoran, R.J.; Lehn, J.-M. Cyclodextrin chemistry. Selective modification of all primary hydroxyl groups of α- and β-cyclodextrins. Helv. Chim. Acta 1978, 61, 2190–2218.
- Letort, S.; Balieu, S.; Erb, W.; Gouhier, G.; Estour, F. Interactions of cyclodextrins and their derivatives with toxic organophosphorus compounds. Beilstein J. Org. Chem. 2016, 12, 204–228.
- Ueno, A.; Breslow, R. Selective sulfonation of a secondary hydroxyl group of β-cyclodextrin. Tetrahedron Lett. 1982, 23, 3451–3454.
- Law, H.; Baussanne, I.; García-Fernández, J.M.; Defaye, J. Regioselective sulfonylation at O-2 of cyclomaltoheptaose with 1-(p-tolylsulfonyl)-(1H)-1,2,4-triazole. Carbohydr. Res. 2003, 338, 451–453.
- Teranishi, K.; Tanabe, S.; Hisamatsu, M.; Yamada, T. Efficient regioselective synthesis of mono-2-O-sulfonyl-cyclodextrins by the combination of sulfonyl imidazole and molecular sieves. J. Carbohydr. Chem. 1998, 17, 489–494.
- Menuel, S.; Doumert, B.; Saitzek, S.; Ponchel, A.; Delevoye, L.; Monflier, E.; Hapiot, F. Selective secondary face modification of cyclodextrins by mechanosynthesis. J. Org. Chem. 2015, 80, 6259–6266.
- Jindrich, J.; Pitha, J.; Lindberg, B.; Seffers, P.; Harata, K. Regioselectivity of alkylation of cyclomaltoheptaose (β-cyclodextrin) and synthesis of its mono-2-O-methyl, -ethyl, -allyl, and -propyl derivatives. Carbohydr. Res. 1995, 266, 75–80.
- Du Roizel, B.; Baltaze, J.-P.; Sinaÿ, P. Diisobutylaluminum-promoted secondary rim selective de-O-methylation of permethylated cyclodextrins. Tetrahedron Lett. 2002, 43, 2371–2373.
- Xiao, S.; Yang, M.; Yu, F.; Zhang, L.; Zhou, D.; Sinaÿ, P.; Zhang, Y. Synthesis of four mono-functionalized α-cyclodextrin derivatives for further confirming DIBAL-H-promoted bis-de-O-methylation mechanism. Tetrahedron 2013, 69, 4053–4060.
- Teranishi, K. Regiospecific 2A,2C-disulfonation of β-cyclodextrin. Tetrahedron Lett. 2000, 41, 933–936.
- Teranishi, K. Practical and convenient modifications of the A,C-secondary hydroxyl face of cyclodextrins. Tetrahedron 2003, 59, 2519–2538.
- Teranishi, K. Regioselective 2A-2D-disulfonylations of cyclodextrins for practical bifunctionalization on the secondary hydroxyl face. J. Incl. Phenom. Macrocycl. Chem. 2002, 44, 313–316.
- Coleman, A.W.; Zhang, P.; Parrot-Lopez, H.; Ling, C.-C.; Miocque, M.; Mascrier, L. The first selective per-tosylation of the secondary OH-2 of β-cyclodextrin. Tetrahedron Lett. 1991, 32, 3997–3998.
- Rong, D.; D’Souza, V.T. A convenient method for functionalization of the 2-position of cyclodextrins. Tetrahedron Lett. 1990, 31, 4275–4278.
- Yu, H.; Teramoto, A.; Fukudome, M.; Xie, R.-G.; Yuan, D.-Q.; Fujita, K. A facile sulfonylation method enabling direct syntheses of per(2-O-sulfonyl)-β-cyclodextrins. Tetrahedron Lett. 2006, 47, 8837–8840.
- Ashton, P.R.; Boyd, S.E.; Gattuso, G.; Hartwell, E.Y.; Koeniger, R.; Spencer, N.; Fraser Stoddart, R. A novel approach to the synthesis of some chemically-modified cyclodextrins. J. Org. Chem. 1995, 60, 3898–3903.
- Dittmann, H.; Scharwächter, K.; König, W.A. Synthesis and silica-based immobilization of monofunctionalized cyclomaltoheptaose derivatives for enantioselective HPLC. Carbohydr. Res. 2000, 324, 75–96.
- Junge, M.; König, W.A. Selectivity tuning of cyclodextrin derivatives by specific substitution. J. Sep. Sci. 2003, 26, 1607–1614.
- Yuan, D.-Q.; Tahara, T.; Chen, W.-H.; Okabe, Y.; Yang, C.; Yagi, Y.; Nogami, Y.; Fukudome, M.; Fujita, K. Functionalization of cyclodextrins via reactions of 2,3-anhydrocyclodextrins. J. Org. Chem. 2003, 68, 9456–9466.
- Wang, Z.; Lu, R. Facile direct acylation and acyl migration of β-cyclodextrin on the secondary hydroxyl face. J. Incl. Phenom. Macrocycl. Chem. 2009, 63, 373–378.
- Fujita, K.; Tahara, T.; Egashira, Y.; Imoto, T.; Koga, T. A complete set of 3A,6X-di-O-sulfonylated α-cyclodextrins. Tetrahedron Lett. 1992, 33, 5385–5388.
- Tian, S.; Forgo, P.; D’Souza, V.T. Selective modification at the 3-position of β-cyclodextrin. Tetrahedron Lett. 1996, 37, 8309–8312.
- Fujita, K.; Nagamura, S.; Imoto, T.; Tahara, T.; Koga, T. Regiospecific sulfonation of secondary hydroxyl groups of alpha-cyclodextrin. Its application to preparation of 2A2B-, 2A2C-, and 2A2D-disulfonates. J. Am. Chem. Soc. 1985, 107, 3233–3235.
- Fujita, K.; Tahara, T.; Yamamura, H.; Imoto, T.; Koga, T.; Mihashi, K. Specific preparation and structure determination of 3A,3C,3E-tri-O-sulfonyl-beta-cyclodextrin. J. Org. Chem. 1990, 55, 877–880.
- Guitet, M.; Adam de Beaumais, S.; Blériot, Y.; Vauzeilles, B.; Zhang, Y.; Ménand, M.; Sollogoub, M. Cyclodextrins selectively modified on both rims using an O-3-debenzylative post-functionalisation, a consequence of the Sorrento meeting. Carbohydr. Res. 2012, 356, 278–281.
- Szejtli, J.; Lipták, A.; Jodál, I.; Fügedi, P.; Nánási, P.; Neszmélyi, A. Synthesis and 13C-NMR spectroscopy of methylated beta-cyclodextrins. Starch-Starke 1980, 32, 165–169.
- Yao, X.; Huanga, P.; Nie, Z. Cyclodextrin-based polymer materials: From controlled synthesis to applications. Progr. Polym. Sci. 2019, 93, 1–35.
- Przybyla, M.A.; Yilmaz, G.; Becer, C.R. Natural cyclodextrins and their derivatives for polymer synthesis. Polym. Chem. 2020, 11, 7582–7602.
- Seidi, F.; Jin, Y.; Xiao, H. Polycyclodextrins: Synthesis, functionalization, and applications. Carbohydr. Polym. 2020, 242, 116277.
- Ohno, K.; Wong, B.; Haddleton, D.M. Synthesis of well-defined cyclodextrin-core star polymers. J. Polym. Sci. A Pol. Chem. 2001, 39, 2206–2214.
- Abdouni, Y.; Yilmaz, G.; Becer, C.R. Sequence controlled polymers with a β-cyclodextrin core. Macromol. Rapid Commun. 2017, 38, 1700501.
- Zhang, M.; Xiong, Q.; Shen, W.; Zhang, Q. Facile synthesis of well-defined cyclodextrin-pendant polymer via ATRP for nanostructure fabrication. RSC Adv. 2014, 4, 30566–30572.
- Diget, J.S.; Städe, L.W.; Nielsen, T.T. Direct synthesis of well-defined zwitterionic cyclodextrin polymers via atom transfer radical polymerization. Eur. Polym. J. 2019, 116, 84–90.
- Perrier, S. 50th Anniversary Perspective: RAFT polymerization—A user guide. Macromolecules 2017, 50, 7433–7447.
- Zhang, L.; Stenzel, M.H. Spherical glycopolymer architectures using RAFT: From stars with a β-cyclodextrin core to thermoresponsive core-shell particles. Aust. J. Chem. 2009, 62, 813–822.
- Koyanagi, K.; Takashima, Y.; Nakamura, T.; Yamaguchi, H.; Harada, A. Radical polymerization by a supramolecular catalyst: Cyclodextrin with a RAFT reagent. Beilstein J. Org. Chem. 2016, 12, 2495–2502.
- Miao, Y.; Zinck, P. Ring-opening polymerization of cyclic esters initiated by cyclodextrins. Polym. Chem. 2012, 3, 1119–1122.
- Gou, P.-F.; Zhu, W.-P.; Xu, N.; Shen, Z.-Q.J. Synthesis and characterization of well-defined cyclodextrin-centered seven-arm star poly(ε-caprolactone)s and amphiphilic star poly(ε-caprolactone-b-ethylene glycol)s. J. Polym. Sci. A Pol. Chem. 2008, 46, 6455–6465.
- Galia, A.; Scialdone, O.; Spanò, T.; Valenti, M.G.; Grignard, B.; Lecomte, P.; Monflier, E.; Tilloy, S.; Rousseau, C. Ring opening polymerization of ε-caprolactone in the presence of wet β-cyclodextrin: Effect of the operative pressure and of water molecules in the β-cyclodextrin cavity. RSC Adv. 2016, 6, 90290–90299.
- Meimoun, J.; Phuphuak, Y.; Miyamachi, R.; Miao, Y.; Bria, M.; Rousseau, C.; Nogueira, G.; Valente, A.; Favrelle-Huret, A.; Zinck, P. Cyclodextrins initiated ring-opening polymerization of lactide using 4-dimethylaminopyridine (DMAP) as catalyst: Study of DMAP/β-CD inclusion complex and access to new structures. Molecules 2022, 27, 1083.
- Faugeras, P.A.; Boëns, B.; Elchinger, P.H.; Brouillette, F.; Montplaisir, D.; Zerrouki, R.; Lucas, R. When cyclodextrins meet click chemistry. Eur. J. Org. Chem. 2012, 2012, 4087–4105.
- Ge, Z.; Xu, J.; Hu, J.; Zhang, Y.; Liu, S. Synthesis and supramolecular self-assembly of stimuli-responsive water-soluble Janus-type heteroarm star copolymers. Soft Matter 2009, 5, 3932–3939.
- Arslan, M.; Sanyal, R.; Sanyal, A. Cyclodextrin embedded covalently crosslinked networks: Synthesis and applications of hydrogels with nano-containers. Polym. Chem. 2020, 11, 615–629.
- Roy, A.; Manna, K.; Dey, S.; Pal, S. Chemical modification of β-cyclodextrin towards hydrogel formation. Carbohydr. Polym. 2023, 306, 120576.
- Iravani, S.; Varma, R.S. Nanosponges for drug delivery and cancer therapy: Recent advances. Nanomaterials 2022, 12, 2440.
- Pawara, S.; Shendea, P.; Trotta, F. Diversity of β-cyclodextrin-based nanosponges for transformation of actives. Int. J. Pharmaceut. 2019, 565, 333–350.
- Krabicová, I.; Appleton, S.L.; Tannous, M.; Hoti, G.; Caldera, F.; Pedrazzo, A.R.; Cecone, C.; Cavalli, R.; Trotta, F. History of cyclodextrin nanosponges. Polymers 2020, 12, 1122.
- Utzeri, G.; Matias, P.M.C.; Murtinho, D.; Valente, A.J. Cyclodextrin-based nanosponges: Overview and opportunities. Front. Chem. 2022, 10, 859406.
- Asela, I.; Donoso-González, O.; Yutronic, N.; Sierpe, R. β- cyclodextrin-based nanosponges functionalized with drugs and gold nanoparticles. Pharmaceutics 2021, 13, 513.
- Guineo-Alvarado, J.; Quilaqueo, M.; Hermosilla, J.; González, S.; Medina, C.; Rolleri, A.; Lim, L.T.; Rubilar, M. Degree of crosslinking in β-cyclodextrin-based nanosponges and their effect on piperine encapsulation. Food Chem. 2021, 340, 128132.
- Sadjadi, S.; Malmir, M.; Heravi, M.M.; Raja, M. Magnetic hybrid of cyclodextrin nanosponge and polyhedral oligomeric silsesquioxane: Efficient catalytic support for immobilization of Pd nanoparticles. Int. J. Biol. Macromol. 2019, 128, 638–647.
- Wadhwa, P.; Vij, M.; Dand, N. Wave-assisted techniques, a greener and quicker alternative to synthesis of cyclodextrin-based nanosponges: A review. Recent Pat. Nanotechnol. 2022.
- Ciesielska, A.; Ciesielski, W.; Girek, B.; Girek, T.; Koziel, K.; Kulawik, D.; Lagiewka, J. Biomedical application of cyclodextrin polymers cross-linked via dianhydrides of carboxylic acids. Appl. Sci. 2020, 10, 8463.
- Rubin Pedrazzo, A.; Caldera, F.; Zanetti, M.; Appleton, S.L.; Dhakar, N.K.; Trotta, F. Mechanochemical green synthesis of hyper-crosslinked cyclodextrin polymers. Beilstein J. Org. Chem. 2020, 16, 1554–1563.
This entry is offline, you can click here to edit this entry!