The epithelial–mesenchymal transition (EMT) is a cellular reprogramming process that occurs during embryonic development and adult tissue homeostasis. This process involves epithelial cells acquiring a mesenchymal phenotype. Epithelial–mesenchymal plasticity (EMP) has become a hot issue in colorectal cancer (CRC) because strong inducers of EMT (such as transforming growth factor β, TGF-β) can initiate EMT and regulate metastasis, micro-environment, and immune system resistance in CRC.
- CRC
- EMT
- MET
- EMP
- clinical prognosis
1. Introduction
Colorectal cancer (CRC) is the second-leading cause of cancer-related death [1][2]. It is expected that in 2023, there will be over 150,000 new instances of CRC in the United States [3][4][5]. In recent years, the rate of CRC has risen in China. CRC can be viewed as an indicator of socioeconomic development, and in countries undergoing significant transitions, the incidence rate tends to increase in tandem with the human development index [2][6].
The complicated and tightly regulated process of epithelial–mesenchymal transition (EMT) may be governed by hundreds of transcription factors and thousands of microRNAs [7]. It is believed that metastatic spread and the development of secondary tumors depend on cancer cell EMT plasticity programs overlaid on intrinsic genetic flaws [8]. The progression and development of CRC are driven by the accumulation of somatic mutations in oncogenes and tumor suppressor genes within colonic stem cells [9].
2. Characteristics of EMT
EMT is a biological reprogramming process that allows epithelial cells to take on a mesenchymal phenotype during embryonic development and adult tissue homeostasis. EMT is crucial for the development of the embryo, the healing of wounds, and the advancement of cancer, as it gives cancer cells aggressive phenotype-associated characteristics. Tight junctions dissolve, apical–basal polarity is disrupted, and the cytoskeletal structure is reorganized in tumor cells after EMT is activated. These physical changes promote cell migration away from the original site, invasion of neighboring tissues, metastasis, survival in the bloodstream, and eventually cause metastases to grow in distant organs [10][11][12][13][14][15][16][17][18][19][20][21].
The EMT program does not operate in a strictly binary manner, and cancer cells seldom complete the entire EMT cycle, which would cause them to become totally mesenchymal. EMT is essential for the development of the embryo, the healing of wounds, and the progression of cancer. This is because EMT confers aggressive phenotype-associated traits on cancer cells, which are necessary for the progression of cancer. The latter of these types of junctions are created by the synthesis of epithelial cell adhesion . E-cadherin maintains the connection between epithelial cells in healthy tissue by acting as a glue. After EMT activation, the expression of mesenchymal markers such as N-cadherin, vimentin, fibroblast-specific protein 1, and fibronectin increased and the expression of epithelial markers such as E-cadherin, ZO-1, and occludin decreased. This causes the epithelial cells to lose their characteristic polygonal, cobblestone morphology (Table 1). Cells undergo a transformation in which they assume a spindle-shaped mesenchymal appearance and have the ability to move [14][18][22][23][24][25].
Table 1. Comparison of differences between epithelial and mesenchymal cells.
|
Feature |
Epithelial Cells |
Mesenchymal Cells |
|
ogy |
Polygonal, pebble-like shape |
Elongated, spindle-shaped [17] |
|
Polarity |
Apical–basal |
Front–back [20] |
|
Invasion ability |
No mobility |
Enhanced movement and raiding capabilities [26] |
|
Cytoskeleton |
Express cytokeratins |
Express vimentin |
|
Intercellular junction |
Tight junctions, adherens junctions, and desmosomes |
Loosely attached to the extracellular matrix [26] |
|
Interactions with extracellular matrix |
Via integrin α6β4 at hemidesmosomes; linked to cytokeratins |
Via β1-containing or β3-containing integrins at adhesion plaques; linked to actin stress fibers [14][20] |
|
Markers |
Junctional proteins, epithelial markers, matrix proteins |
Matrix proteins, proteases, mesenchymal markers [27] |
It is not shocking that EMT has been linked to the spread of tumor cells because it increases cell mobility [16]. EMT works as a dynamic program that alternates between alternate epithelial and mesenchymal phases to produce a variety of cells with different phenotypes. In addition, the fact that the execution of the EMT program can be influenced by the environmental signals that are experienced by cells as well as the pre-existing differentiation lineages of these cells means that EMT cannot be viewed as a singular and stereotypical program. Instead, EMT can be described as a collection of programs that share many elements in common.
EMT is a reversible biological process that temporarily transforms epithelial cells into a paramesenchymal state. mesenchymal–epithelial transition (MET), a reversal process, allows the extra mesenchymal cells to shift back to the epithelial state [14]. Numerous investigations have pointed to the necessity of reversing EMT as a prerequisite for successful metastatic colonization [13][14][18][24][28][29][30][31]. The environment has a significant impact on the occurrence of EMT and/or MET, with cell type and tissue type playing the most significant roles [31]. In the intricate progression from primary tumor to metastasis, cancer cells must adapt to shifting and frequently unfavorable environmental conditions. This tumor cell plasticity is demonstrated by the reversible transition from differentiated to undifferentiated or partially EMT-associated cancer cell phenotypes [32]. Human CRC cell clusters in an E/M state contribute to metastatic seeding (Figure 1), and the loss of E-cadherin expression during culture frequently induces EMT in pure mesenchymal tumor cells. Thus, the characteristics of local invasion and tumor initiation are attained [33].
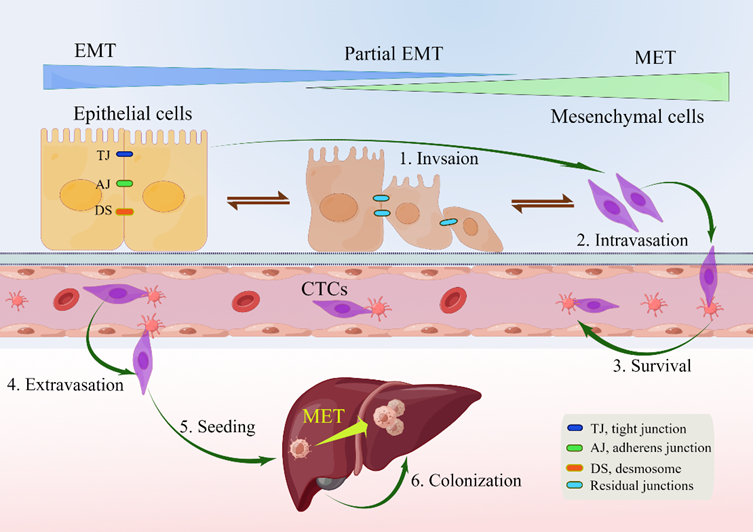
Figure 1. Depiction of EMP and invasion–metastasis cascades in CRC cells. Along the entire EMT spectrum, CRC cells have different degrees of growth and metastasis. Cells can move along these two states with flexibility since EMT and MET are nonbinary reversible processes. Malignant cells were those that exhibited more epithelial characteristics and less mesenchymal transition. Primary CRC cells undergoing EMT undergo a series of intermolecular changes that lead to loss of intercellular adhesion, including dissolution of intercellular junctions, namely tight junctions (dark blue), adherens junctions (green), and desmosomes (red orange); through this process, cells acquire a mesenchymal phenotype that promotes local migration and invasion. The cells then enter the blood vessels (intravasation), survive in the blood vessels as circulating tumor cells, and leave the blood vessels (extravasation) to seed and colonize the parenchyma of the liver. After seeding locally in the liver, cancer cells undergoing EMT can redifferentiate to an epithelial phenotype through MET, a step that facilitates cell colonization in the liver and development of local metastases. Abbreviations: EMP, epithelial–mesenchymal plasticity; CTC, circulating tumor cells; EMT, epithelial–mesenchymal transition; MET, mesenchymal–epithelial transition.
3. EMT in CRC
Partial EMT is present in more than 90% of human CRC cell lines, which is a condition that encourages the formation of cell clusters during CRC spread. EMT is therefore a prospective target for preventing original cancers from becoming aggressive or for preventing metastasis and recurrence following tumor resection. The adaptability and variety of the different pathways involved make it difficult to develop medications that specifically target EMT. To combat drug resistance in metastatic CRC, however, promising therapeutic approaches include: (1) combining EMT inhibitors with conventional chemotherapy medicines; and (2) employing EMT inhibitors in adjuvant therapy to decrease tumor resection relapse [15].
By downregulating E-cadherin and other components of epithelial junctions, CRC cells isolate themselves from one another and from nearby normal or cancer cells in the initial step of the invasion–metastasis cascade. They subsequently penetrate through the underlying stroma and destroy the basement membrane by secreting or locally activating matrix metalloproteinases (MMPs) and urokinase plasminogen activator (u-PA). Proangiogenic factors, including VEGF and other proangiogenic cytokines, are released as a result of the pericellular matrix and extracellular matrix (ECM) degrading. These factors encourage the growth of new lymphatic and blood capillaries, which aids in the infiltration of tumor cells into the bloodstream. Because of this, tumor cells can either enter blood arteries directly or indirectly through colon-lymphatic channels [15].
4. Mechanism of EMT in CRC
Transcriptional regulation, post-translational control, epigenetic modification, and noncoding RNA-mediated regulation are some of the complex network components that closely regulate EMT. Snail, Twist, Zeb, and other EMT-related transcription factors are expressed at the post-transcriptional and post-translational levels under the regulation of many signaling pathways (Figure 2). These pathways also regulate the downstream transcriptional network, which further controls the biological impacts of EMT, in conjunction with other epigenetic variables. They suppress the production of genes that uphold the epithelial state and promote the mesenchymal state [10][13][14][16][18][20][24][32][34][35].
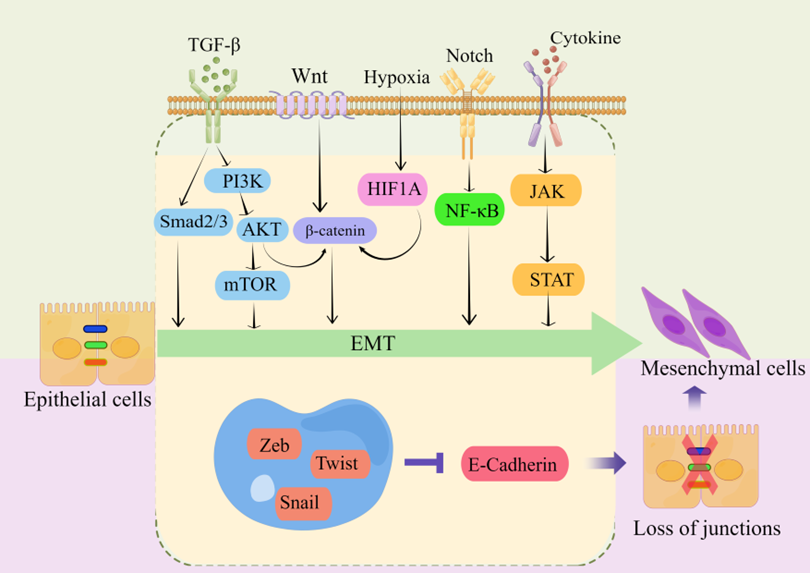
Figure 2. regulating EMT-TFs during EMT development in CRC. There are several EMT-TFs (such as ZEB, SNAIL, and TWIST) that cooperate with signaling pathways in cells, and under the stimulation of factors such as TGF-β, Wnt, Notch, hypoxia, etc., repress genes related to epithelial status (such as E-cadherin), which leads to the loss of intercellular connections and allows epithelial cells to differentiate into a mesenchymal state. These transcription factors are pleiotropic, inducing a transition to a mesenchymal or partially mesenchymal cellular state. Abbreviations: EMT, epithelial–mesenchymal transition; PI3K, phosphatidylinositol-3-kinase; AKT, protein kinase B; mTOR, mammalian target of rapamycin; HIF1A, hypoxia inducible factor 1 subunit alpha; NF-κB, nuclear factor kappa B; JAK, janus kinase; STAT, signal transducer and activator of transcription; ZEB, zinc finger E-box-binding homeobox.
Multiple signaling pathways, including TGF-β/SMAD, WNT/β-catenin, Notch, and receptor tyrosine kinase, can be activated by extracellular stimuli from the tumor microenvironment. These signaling pathways induce a core set of EMT transcription factors to initiate the EMT program in tumor cells [39]. The EMT program directed by EMT-TFs can bestow various qualities essential to the malignant evolution of tumor cells, including tumor-initiating traits, motility, dissemination capability, and greater resistance to frequently employed chemotherapeutic medicines [14][40]. Even though a sizeable majority of the research conducted on EMT focuses on transcription factor networks, EMT can also be controlled by post-translational modifications, microRNAs, long noncoding RNAs, post-transcriptional alterations, epigenetic regulators, and alternative splicing. In order to produce these highly flexible cellular states, various levels of regulation do not serve as independent mediators; rather, they constitute a highly interconnected network.
5. The Clinical Role of EMT
EMT has a significant impact on tumor development, metastasis, and medication resistance in CRC. There is growing proof that EMT indicators can act as outcome predictors and possible treatment targets in CRC from preclinical and early clinical research [10]. E-cadherin downregulation, which causes adhesion junctions to become unstable, is a crucial component in EMT. In stage III CRC, a poor prognosis is linked to loss of E-cadherin expression. It has been discovered that CRC patients have an elevated risk of cancer recurrence and a decreased chance of survival due to aberrant regulation of transcription factors connected to EMT and mesenchymal markers. According to earlier research, the aggressiveness, metastasis, and poor prognosis of CRC are all connected to the overexpression of transcription factors relevant to EMT, such as SNAIL, SLUG, TWIST, and ZEB [43]. Vimentin processes mechanical feedback and controls the dynamics of microtubule and actin networks to aid in the promotion of cell migration. Therefore, there is little question that vimentin’s enhanced expression encourages CRC to invade [10].
EMT caused by elevated mesenchymal gene expression in epithelial tumors is a sign of a poor prognosis in CRC. The expression patterns of both mesenchymal cells present in the tumor microenvironment (TME) and epithelial cancer cells are reflected in the transcriptome of tumor tissues. In the transcriptome-based CRC data, TME cells, particularly cancer-associated fibroblasts (CAFs), were primarily responsible for the expression of mesenchymal genes as opposed to cancer cells [41].
The majority of invasive epithelial carcinoma cells go through EMT. Cancer cells in the mesenchymal and intermediate phases group together with other cells in the microenvironment to form clumps, invade blood arteries, and develop into CTCs. CTCs facilitate the spread of CRC to the liver, lungs, and lymph nodes. CTCs isolated from orthotopic CRC xenograft models generated organoids (CTCs-derived organoids, CTCDOs). CTCDO exhibited a mixed EMT state and increased expression of stemness-related markers. Functionally, CTCDOs showed higher migration/invasion abilities and different responses to pathway-targeting drugs compared with xenograft-derived organoids (XDOs) [19][42]. It has also been reported that adjuvant therapy for colon cancer patients guided by circulating tumor DNA (ctDNA) is beneficial [43].
6. Targeting EMT in the Treatment of CRC
The prognosis of patients with CRC varies, with tumors on the right side having the worst prognosis according to the site of the primary tumor, so screening and early diagnosis strategies for these patients are particularly important [44]. It is difficult to develop clinical routes that target EMT modulators, effectors, or inducers because of the variability and plasticity of the several pathways involved. The utilization of EMT components as therapeutic targets is further complicated by spillover effects between pathways [10]. It is therefore doubtful that targeting a single EMT receptor will be successful due to the redundancy of many pathways; nevertheless, targeting upstream transcription factors may have a more significant impact.
From a therapeutic perspective, it would be beneficial to reverse the process of EMT, i.e., induce MET, given the critical role of the EMT program in the multiple malignant features of cancer. Cancer cells in a CSC state with an active EMT program will be compelled to undergo differentiation into non-CSCs and revert to epithelial features as a result, losing their increased proliferative potential and resistance to diverse treatments [20]. Inactivation of EMT and induction of MET are necessary for efficient metastatic colonization and growth at a distance. In this context, therapeutic promotion of MET may accelerate metastasis of disseminating cells. Therefore, the precise treatment period needs to be carefully defined. Loss of E-cadherin is a key feature of EMT, and restoring its expression may be considered a promising approach to suppress metastasis [10].
It has been shown that TGF-β can promote EMT through both SMAD-dependent and -independent mechanisms. Therefore, therapeutic strategies that target these signaling pathways through TGF-β may be effective in stopping cancer cells from growing invasively and from spreading [36]. Targeting CMS4-like TGF-β-activated CRC, clinical trials have started on the first treatment combination, opening the door to overcome the tumor microenvironmental interaction that maintains the mesenchymal phenotype[45].
Multiple lines of evidence indicate that EMT is an epigenetic process independent of alterations in the DNA sequence of normal and tumor cells. The expression of this program and its comprehensive impact on tumor biology cannot be determined by sequencing the genomes of cancer cells, as is the case with many other biological programs [14]. EMT appears to be a crucial consideration for improving and refining the current clinical classification of CRC and better stratifying patients for prognosis and therapy. However, the lack of reliable and robust biomarkers continues to restrict its application.
7. Conclusions
When considered as a whole, EMT plays a crucial part in the progression of CRC and is a prospective target for preventing colon cancer from acquiring invasiveness or avoiding recurrence and metastasis after its excision. Both of these outcomes are associated with the progression of the disease. Therefore, modification of the several pathways that target EMT could be a viable therapeutic strategy for CRC.
In clinical settings, the identification and development of EMT-associated biomarkers has been hampered by the difficulties of correctly recognizing the EMT process at the morphological level. However, the development of liquid biopsy, which includes CTCs and ctDNA, has opened up a new door in the early detection of CRC, as well as in the monitoring of the disease, the treatment response, and the development of drug resistance.
In the end, it will be important to identify a general consensus of robust, validated biomarkers in order to enhance the detection of, treatment for, and prognosis of CRC.
This entry is adapted from the peer-reviewed paper 10.3390/ijms241914815
References
- Siegel, R. L.; Miller, K. D.; Wagle, N. S., et al., Cancer statistics, 2023[J]. CA Cancer J Clin, 2023, 73: 17-48
- Sung, H.; Ferlay, J.; Siegel, R. L., et al., Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries[J]. CA Cancer J Clin, 2021, 71: 209-249
- Wong, S. H.; Yu, J., Gut microbiota in colorectal cancer: mechanisms of action and clinical applications[J]. Nat Rev Gastroenterol Hepatol, 2019, 16: 690-704
- Ciardiello, F.; Ciardiello, D.; Martini, G., et al., Clinical management of metastatic colorectal cancer in the era of precision medicine[J]. CA Cancer J Clin, 2022, 72: 372-401
- Patel, S. G.; Karlitz, J. J.; Yen, T., et al., The rising tide of early-onset colorectal cancer: a comprehensive review of epidemiology, clinical features, biology, risk factors, prevention, and early detection[J]. Lancet Gastroenterol Hepatol, 2022, 7: 262-274
- Qu, R.; Ma, Y.; Zhang, Z., et al., Increasing burden of colorectal cancer in China[J]. Lancet Gastroenterol Hepatol, 2022, 7: 700
- Bracken, C. P.; Goodall, G. J., The many regulators of epithelial-mesenchymal transition[J]. Nat Rev Mol Cell Biol, 2022, 23: 89-90
- LeBleu, V. S.; Thiery, J. P., The Continuing Search for Causality between Epithelial-to-Mesenchymal Transition and the Metastatic Fitness of Carcinoma Cells[J]. Cancer Res, 2022, 82: 1467-1469
- Sedlak, J. C.; Yilmaz Ö, H.; Roper, J., Metabolism and Colorectal Cancer[J]. Annu Rev Pathol, 2023, 18: 467-492
- Zhang, N.; Ng, A. S.; Cai, S., et al., Novel therapeutic strategies: targeting epithelial-mesenchymal transition in colorectal cancer[J]. Lancet Oncol, 2021, 22: e358-e368
- Singh, M.; Yelle, N.; Venugopal, C., et al., EMT: Mechanisms and therapeutic implications[J]. Pharmacol Ther, 2018, 182: 80-94
- Stemmler, M. P.; Eccles, R. L.; Brabletz, S., et al., Non-redundant functions of EMT transcription factors[J]. Nat Cell Biol, 2019, 21: 102-112
- Lamouille, S.; Xu, J.; Derynck, R., Molecular mechanisms of epithelial-mesenchymal transition[J]. Nat Rev Mol Cell Biol, 2014, 15: 178-96
- Dongre, A.; Weinberg, R. A., New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer[J]. Nat Rev Mol Cell Biol, 2019, 20: 69-84
- Shin, A. E.; Giancotti, F. G.; Rustgi, A. K., Metastatic colorectal cancer: mechanisms and emerging therapeutics[J]. Trends Pharmacol Sci, 2023, 44: 222-236
- Aiello, N. M.; Kang, Y., Context-dependent EMT programs in cancer metastasis[J]. J Exp Med, 2019, 216: 1016-1026
- Ye, X.; Weinberg, R. A., Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression[J]. Trends Cell Biol, 2015, 25: 675-686
- Lambert, A. W.; Weinberg, R. A., Linking EMT programmes to normal and neoplastic epithelial stem cells[J]. Nat Rev Cancer, 2021, 21: 325-338
- Morin, C.; Moyret-Lalle, C.; Mertani, H. C., et al., Heterogeneity and dynamic of EMT through the plasticity of ribosome and mRNA translation[J]. Biochim Biophys Acta Rev Cancer, 2022, 1877: 188718
- Shibue, T.; Weinberg, R. A., EMT, CSCs, and drug resistance: the mechanistic link and clinical implications[J]. Nat Rev Clin Oncol, 2017, 14: 611-629
- Derynck, R.; Weinberg, R. A., EMT and Cancer: More Than Meets the Eye[J]. Dev Cell, 2019, 49: 313-316
- O’Driscoll L. When E-cadherin becomes unstuck in cancer[J]. New England Journal of Medicine, 2020, 383(9): 871-873.
- Celesti, G.; Di Caro, G.; Bianchi, P., et al., Presence of Twist1-positive neoplastic cells in the stroma of chromosome-unstable colorectal tumors[J]. Gastroenterology, 2013, 145: 647-57.e15
- Huang, Y.; Hong, W.; Wei, X., The molecular mechanisms and therapeutic strategies of EMT in tumor progression and metastasis[J]. J Hematol Oncol, 2022, 15: 129
- Fiori, M. E.; Di Franco, S.; Villanova, L., et al., Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance[J]. Mol Cancer, 2019, 18: 70
- Tiwari, N.; Gheldof, A.; Tatari, M., et al., EMT as the ultimate survival mechanism of cancer cells[J]. Semin Cancer Biol, 2012, 22: 194-207
- Schleimer, R. P., Immunopathogenesis of Chronic Rhinosinusitis and Nasal Polyposis[J]. Annu Rev Pathol, 2017, 12: 331-357
- Brabletz, T., EMT and MET in metastasis: where are the cancer stem cells?[J]. Cancer Cell, 2012, 22: 699-701
- Shen, M.; Kang, Y., Role Reversal: A Pro-metastatic Function of E-Cadherin[J]. Dev Cell, 2019, 51: 417-419
- Liu, H.; Li, D.; Sun, L., et al., Interaction of lncRNA MIR100HG with hnRNPA2B1 facilitates m(6)A-dependent stabilization of TCF7L2 mRNA and colorectal cancer progression[J]. Mol Cancer, 2022, 21: 74
- Bakir, B.; Chiarella, A. M.; Pitarresi, J. R., et al., EMT, MET, Plasticity, and Tumor Metastasis[J]. Trends Cell Biol, 2020, 30: 764-776
- Brabletz, T.; Kalluri, R.; Nieto, M. A., et al., EMT in cancer[J]. Nat Rev Cancer, 2018, 18: 128-134
- Mizukoshi, K.; Okazawa, Y.; Haeno, H., et al., Metastatic seeding of human colon cancer cell clusters expressing the hybrid epithelial/mesenchymal state[J]. Int J Cancer, 2020, 146: 2547-2562
- Su, J.; Morgani, S. M.; David, C. J., et al., TGF-β orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1[J]. Nature, 2020, 577: 566-571
- Mittal, V., Epithelial Mesenchymal Transition in Tumor Metastasis[J]. Annu Rev Pathol, 2018, 13: 395-412
- Bahrami, A.; Majeed, M.; Sahebkar, A., Curcumin: a potent agent to reverse epithelial-to-mesenchymal transition[J]. Cell Oncol (Dordr), 2019, 42: 405-421
- Chong, W.; Zhu, X.; Ren, H., et al., Integrated multi-omics characterization of KRAS mutant colorectal cancer[J]. Theranostics, 2022, 12: 5138-5154
- Pattabiraman, D. R.; Weinberg, R. A., Tackling the cancer stem cells - what challenges do they pose?[J]. Nat Rev Drug Discov, 2014, 13: 497-512
- Khanbabaei, H.; Ebrahimi, S.; García-Rodríguez, J. L., et al., Non-coding RNAs and epithelial mesenchymal transition in cancer: molecular mechanisms and clinical implications[J]. J Exp Clin Cancer Res, 2022, 41: 278
- Verstappe, J.; Berx, G., A role for partial epithelial-to-mesenchymal transition in enabling stemness in homeostasis and cancer[J]. Semin Cancer Biol, 2023, 90: 15-28
- Tauriello, D. V. F.; Batlle, E., Targeting the Microenvironment in Advanced Colorectal Cancer[J]. Trends Cancer, 2016, 2: 495-504
- De Angelis, M. L.; Francescangeli, F.; Nicolazzo, C., et al., An organoid model of colorectal circulating tumor cells with stem cell features, hybrid EMT state and distinctive therapy response profile[J]. J Exp Clin Cancer Res, 2022, 41: 86
- Hindié, E., Circulating Tumor DNA Guiding Adjuvant Therapy in Colon Cancer[J]. N Engl J Med, 2022, 387: 759
- Gogenur, I.; Qvortrup, C., Colorectal cancer screening in Europe: what are the next steps?[J]. Lancet Oncol, 2021, 22: 898-899
- Rajamäki, K.; Taira, A.; Katainen, R., et al., Genetic and Epigenetic Characteristics of Inflammatory Bowel Disease-Associated Colorectal Cancer[J]. Gastroenterology, 2021, 161: 592-607
- Rajamäki, K.; Taira, A.; Katainen, R., et al., Genetic and Epigenetic Characteristics of Inflammatory Bowel Disease-Associated Colorectal Cancer[J]. Gastroenterology, 2021, 161: 592-607