Complement Component 1q (C1q), an initiating recognition molecule of the classical complement pathway, can interact with a variety of ligands and perform a range of functions in physiological and pathophysiological conditions of the central nervous system (CNS). Beyond its established roles in CNS growth, development, and bodily immunization, a novel facet of C1q’s functionality has been freshly unveiled within the intricate tapestry of neuropathological pathways that underpin neurodegenerative disorders and traumatic brain injury (TBI). This revelation has cast a spotlight on C1q as a prospective therapeutic avenue for safeguarding neuronal well-being or for retarding the progression of neurodegenerative maladies.
1. Alzheimer’s Disease
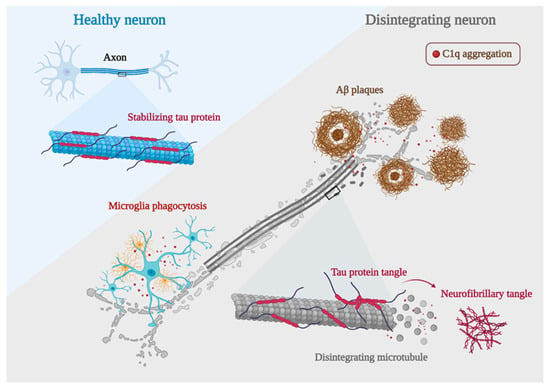
In a series of experiments, excessive complement-mediated synapse pruning was found to be involved in the process of forgetting in AD. Region-specific loss of synaptic salience is a more potent contributor to cognitive decline in AD than the hallmark features of AD, Aβ plaques, and Tau protein hyperphosphorylation [
1]. Unlike the state of synapses requiring proper pruning during growth and development, it has been shown that the number of synapses is significantly reduced in patients with early AD. The number of synapses in 75% of patients with mild cognitive impairment was lower than the average of normal individuals, and the number of synapses correlated significantly with the cognitive–behavioral status of AD patients [
97,
98,
99]. Similarly, synapses in the temporal cortex are reduced by 38% and in the frontal cortex by 14% in AD patients compared with normal subjects [
98]. AD mice lacking C1q display reduced synapse loss, supporting a role for C1q in mediating synapse removal [
2]. At the end of AD, synapse number decreases positively with the degree of cognitive–behavioral impairment in AD patients [
99]. Interestingly, C1q can be observed colocalized with either pre-synaptic or post-synaptic markers in animal models of aging-related diseases, including AD. Accordingly, the upregulation of C1q-tagged synapses is also proved in AD and other neurogenerative disorders-induced cognitive loss. Conversely, the knockdown or blockade of C1q in mouse models of AD has been shown to protect synapses and prevent cognitive impairment, suggesting the detrimental influence of C1q in synaptic loss, and even C1q-labeled synaptic loss may directly contribute to the worsening of AD.
AD is characterized by synaptic dysfunction and neurodegeneration, which are often caused by the deposition of Aβ plaques and neurofibrillary tangles [
100]. The deposition of Aβ plaques triggers a series of chain reactions that lead to intracellular Tau protein misfolding and assembly, which subsequently allows the spread of the lesion throughout the neural circuit as well as the cortex, ultimately leading to neurological failure and cognitive decline. C1q plays an important role in this. It has been shown that blocking C1q activation by genetic or antibody-mediated means can attenuate the toxic effects of Aβ and hyperphosphorylated Tau on synapses [
101]. This provides another direct evidence for a deleterious effect of C1q during the process of AD (
Figure 4).
Figure 4. AD development mechanisms. Neurons with C1q aggregation show microglia engulfing healthy synapses, Aβ clumping forming plaques, hyperphosphorylated Tau proteins forming neurofibrillary tangles, and a significant decrease in their ability to bind microtubules due to Tau protein hyperphosphorylation, leading to microtubule disintegration and ultimately worsening of AD development (created with
BioRender.com accessed on 12 October 2022).
1.1. C1q and Aβ in AD
The major component of amyloid plaques is Aβ, a peptide with 39 to 43 amino acids derived from amyloid b protein precursor (APP) [
102]. Studies have shown that the imbalance between the production and clearance of Aβ and related Aβ peptides plays a fundamental role in the pathogenesis of AD [
103,
104]. In vitro experiments have shown that C1q interacts with Aβ through its A-chain residues 14–26 [
104,
105,
106,
107]. The complement component C1q has nearly 100% co-localization of Aβ in humans with AD and in mouse models of AD [
108]. Sections of the Aβ-treated hippocampus showed a significant increase of C1q in the hippocampus [
109]. When soluble Aβ oligomers were injected into the lateral ventricles of WT mice, Aβ oligomers were found to induce C1q deposition [
1]. Similarly, when J20 mice were injected with a γ-secretase inhibitor that rapidly reduced Aβ production, it significantly reduced soluble Aβ levels in mice with a corresponding reduction in C1q deposition. When C1q knockdown was followed by Aβ injection, the synaptic loss induced by Aβ was significantly reduced [
2]. The use of anti-C1q antibodies similarly prevented Aβ-induced synaptic loss in mice [
1], suggesting that C1q is required for Aβ-induced synaptic loss in vivo. Notably, C1q knockdown does not affect Aβ deposition [
110], so C1q may function downstream of Aβ. Aβ appears abnormally as early as 20 years before the onset of overt clinical symptoms [
111]. Aβ deposition is the beginning of neurodegenerative lesions, but the accumulation of hyperphosphorylated Tau proteins is the main driver of the deteriorating pathological process.
1.2. C1q and Tau in AD
In AD and other Tau lesions, Tau aggregates in an abnormally phosphorylated form in the torso region of neurons and can localize to synapses, where it disrupts synaptic plasticity and leads to synaptic loss [
112]. Positron emission tomography (PET) imaging targeting Aβ and Tau has unveiled a consequential relationship: the velocity of amyloid aggregation forecasts the advent of Tau accumulation, which in turn heralds the initiation of cognitive decline [
113]. In AD, Tau protein aggregation may begin in the entorhinal cortex and then propagate to the hippocampus, as well as within the limbic cortex, reflecting the progression of AD patients from asymptomatic, mildly symptomatic, to full dementia [
114]. It has been shown that hyperphosphorylated Tau protein induces more C1q aggregation at the synapse than Aβ plaques [
101]. In the mouse model, increased Tau phosphorylation and accumulation were accompanied by a dose-dependent increase in C1q [
115]. It has been shown that knockout of the granule protein precursor gene PGRN significantly reduces Aβ plaque production, but deletion of PGRN enhances C1q deposition at the synapse while increasing the accumulation of hyperphosphorylated Tau protein in the hippocampus [
116]. Interestingly, knockdown of the transmembrane immune signaling adaptor TYROBP showed opposite results, where knockdown of TYROBP resulted in a significant reduction in C1q and improvement in memory cognition impairment, hyperphosphorylated Tau protein was not reduced by the decrease in C1q, but instead, its spread was further expanded and Tau protein phosphorylation levels were increased. Suggesting that when there are multiple competing effects occurring simultaneously, the deleterious effects of increased Tau protein phosphorylation levels and diffusion can be overcome as long as the beneficial effect of a significant decrease in C1q is large enough [
117]. It may give insights into the target role of C1q in regulating the progression of AD pathology and cognitive loss.
2. Parkinson Disease
PD is clinically characterized by an akinetic rigid syndrome related to reduced. This syndrome is entwined with a decline in dopamine levels within the striatum, arising from the gradual demise of terminals belonging to degenerating neuromelanin-containing dopaminergic neurons in the substantia nigra pars compacta [
118]. A handful of investigations have scrutinized the complement system within the context of the PD brain. The steady manifestation of C1q was discernible only within microglial cells spanning the cerebral expanse. After MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) exposure, there was an early and temporary elevation in microglial C1q expression within the substantia nigra and striatum, as unveiled through techniques of immunohistochemistry and in situ hybridization. Notably, Rozemuller and collaborators found no immunostaining indicative of C1q within cortical Lewy bodies [
119]. Concurrently, mice devoid of the C1q protein exhibited no substantial differences in terms of the loss of nigral dopaminergic neurons, striatal dopaminergic fibers, and dopamine levels induced by MPTP in comparison to their control counterparts [
120,
121]. This shows that C1q is not a major contributor to cognitive impairment in PD. Simultaneously, within the substantia nigra pars compacta (SNc) of PD cases, there manifested an augmented accumulation of extracellular neuromelanin within the tissue, a manifestation arising from the degeneration of dopaminergic neurons. In this milieu, neuromelanin granules and fragments from deteriorated neurons appeared to be tagged by C1q, thus becoming subject to phagocytosis by C1q-positive microglia and macrophages situated both within the tissue and around perivascular spaces. Notably, cells bearing neuromelanin and C1q also adhered to the inner surfaces of blood vessels in the SNc in the context of PD [
8]. Hence, microglia demonstrate the capability to engulf and eliminate cellular detritus emanating from degenerating neurons within the SNc, effectively orchestrating this process through a pathway facilitated by C1q, a phenomenon that occurs within the context of PD. Although C1q may not play a direct pathological role in PD, it can affect the disease process through microglia phagocytosis, etc.
3. Huntington’s Disease
HD is an autosomal-dominant neurodegenerative disorder characterized by a relentless progression, culminating in targeted neuronal attrition and impairment, predominantly within the striatal and cortical regions [
122]. Within the striatal milieu of HD, a convergence was observed wherein neurons, myelin, and astrocytes demonstrated a spatial overlap with C1q. In contrast, no C1q was found in the normal striatum. In normal control brains, the abundance of C1q mRNA ranged from 2 to 5 times lower when juxtaposed with the levels identified in the striatum affected by HD. The course of HD is marked by a neuroinflammatory progression orchestrated by the activation of microglia within the cerebral domain [
123]. Astrogliosis and microgliosis were apparent in all caudate and internal capsule samples from individuals with HD, a phenomenon absent in normal brain tissue. Microglia of the M1 phenotype within the HD context produced C1q, which was subsequently triggered on neuronal membranes. This dual action of C1q not only facilitated neuronal necrosis but also contributed to proinflammatory activities [
124]. Meanwhile, it has been reported that the secretion of cytokines C1q upon M1 microglial activation can induce the generation of reactive A1 astrocytes at neuronal structures, which play a major role in brain motor coordination [
125,
126]. These intricate processes are believed to precipitate neurodegenerative events within the brain, ultimately giving rise to the motor dysfunctions that become manifest in the later stages of this neurological affliction. Kaempferol, a natural antioxidant found in vegetables and fruits consumed as part of human nutrition, has exhibited the capacity to forestall the proteolytic activation of complement C1q protein and the subsequent emergence of reactive A1 astrocytes. This phenomenon has been observed in the context of 3-nitro propionic acid-induced injury within the striatum and hippocampus. Cognitive–behavioral deficits in experimental animals significantly improved when microglia secretion of C1q was reduced in an animal model of HD [
127].
4. Traumatic Brain Injury
TBI emerges as the most potent environmental catalyst in the emergence of AD and other neurodegenerative disorders linked to dementia. The initial trauma sustained by the brain impairs the integrity of the BBB, consequently permitting the infiltration of peripheral circulating macrophages into the cerebral milieu. This occurrence subsequently accentuates the inflammatory response [
128]. The prevailing notion suggests that a transition between the M1 and M2 microglial phenotypes transpires within the framework of TBI. However, it appears that there exists a proclivity towards favoring the M1 phenotype over the M2 phenotype in the context of TBI-associated secondary injury [
129]. In pathological scenarios encompassing TBI, there is substantiated evidence suggesting that C1q plays a contributory role in steering a shift toward the M1 phenotype [
78]. In tandem with the direct harm incited by M1 microglia, the C1q they release can also instigate the activation of astrocytes [
130]. A prominent constituent of the inflammatory pathway, the complement system, often escapes notice, yet it too undergoes activation as an integral facet of the neuroinflammatory rejoinder in TBI [
131]. The intrinsic complement system within the CNS undergoes activation, with this activation further amplified by an influx of complement components from the bloodstream, facilitated by the disruption of the BBB. In parallel, certain investigations have demonstrated a noteworthy accumulation of C1q on synapses located in the hippocampus. This accumulation aligns temporally with the loss of synapses 30 days after the injury. Significantly, both genetic interventions and the implementation of pharmacological measures to obstruct the complement pathway yielded the prevention of memory deficits in aged animals subjected to injury [
132,
133]. Therefore, strategically focusing on the modulation of C1q emerges as a substantial avenue for potential clinical intervention after TBI within the aging demographic (
Table 2).
Table 2. Characteristics of neurodegenerative diseases.
| Neurodegenerative Diseases |
Animal/Cellular Model |
Characteristics |
C1q Effects |
Ref. |
| AD |
Tg2576 animals (APP) with C1q-deficient mice |
Aβ plaques |
Aβ co-localizes with C1q |
[2,108,109] |
| |
PS19 mice overexpressing the P301S mutant of human Tau/Hek cells |
Tau protein misfolding and assembly |
Tau protein co-localizes with C1q |
[113,114,115] |
| |
Tg2576 animals (APP) with C1q-deficient mice |
Synapse elimination |
Synapse co-localizes with C1q |
[2,3,62] |
| PD |
- |
Depigmentation of the substantia nigra and locus coeruleus |
C1q was restricted to microglia throughout the brain |
[118] |
| |
Autopsies from PD patients |
Neuronal loss in the pars compacta of the substantia nigra |
|
[8,120] |
| HD |
Early HD patients |
CAG trinucleotide repeat expansion in the huntingtin gene on chromosome 4 |
C1q produced locally by M1-type microglia is activated on the membranes of neurons |
[124,134] |
| TBI |
Sections of brains obtained at autopsy from 25 cases following closed TBI |
Traumatic brain injury disrupts the BBB |
C1q prompts the transformation of M2-type microglia into M1-type microglia and enhances complement system activation |
[78,131] |
This entry is adapted from the peer-reviewed paper 10.3390/foods12193580