Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Insulin-like growth factor 1 (IGF1) is a peptide growth factor with important functions in multiple aspects of growth, development and metabolism. The biological actions of IGF1 are mediated by the IGF1 receptor (IGF1R), a cell-surface protein that is evolutionarily related to the insulin receptor (InsR).
- insulin-like growth factor-1 (IGF1)
- IGF1 receptor
- MAPK
- nuclear translocation
- p53
1. Introduction
Insulin-like growth factor-1 (IGF1) was discovered in the late 1950s, when the mechanisms of action of growth hormone (GH, or somatotropin) were investigated in animal models [1]. These early studies provided evidence that sulfate incorporation into cartilage was not a direct action of GH, but, rather an indirect process that involves the biosynthesis of a serum-borne mediator termed ‘sulfation factor’, which is directly responsible for the anabolic activities of GH. The catalogue of activities of the sulfation factor extended in the following years to include stimulation of proline incorporation into collagen, uridine into RNA, thymidine into DNA, etc. Moreover, the huge span of activities associated with the sulfation factor was substantiated by studies showing that its growth-promoting actions were not restricted to cartilage but, in fact, involved additional tissues and organs, including muscle, adipose tissue, brain, etc [2].
The term ‘somatomedin’ was subsequently coined to emphasize the fact that this serum factor mediates the effects of somatotropin at somatic target organs [3][4]. Further biochemical analyses led to the identification of two tightly linked peptides with growth-promoting functions in the serum fraction [5][6]. These peptides were shown to display insulin-like activities in the diaphragm and to stimulate glucose incorporation in fat. However, the insulin-like activities of these molecules were not suppressed by antibodies against insulin [6][7][8]. Hence, the factors that were initially referred to as sulfation factor, somatomedin, and ‘NSILA’ (non-suppressible insulin-like activity) were eventually termed insulin-like growth factor-1 and insulin-like growth factor-2 (IGF1 and IGF2) [9][10]. A recent article by Miller et al. [11] summarized the history of the IGF system.
The biological actions of IGF1 and IGF2 are transduced by a group of transmembrane receptors that includes the insulin (InsR), IGF1 (IGF1R), and IGF2/mannose-6-phosphate (M6P) receptors [12][13][14][15]. While IGF1R is the primary mediator of IGF1/2 action, the InsR and IGF2/M6 receptor are also capable of mediating some of these activities, though with reduced affinities [16][17][18]. The IGF1R is linked to a series of intracellular pathways that includes the RAS-MAPK and PI3K-AKT signaling cascades [19]. The biological actions of the IGFs are moderated by a group of at least six IGF-binding proteins (IGFBP) that are produced by most tissues and are present in variable amounts in the circulation and extracellular compartments [20]. The IGFBPs are capable of inhibiting or enhancing IGF effects in a tissue-specific manner. Furthermore, various IGFBPs, most notably IGFBP3, also display ligand-independent effects [21][22]. A schematic representation of the major components of the IGF family is shown in Figure 1.
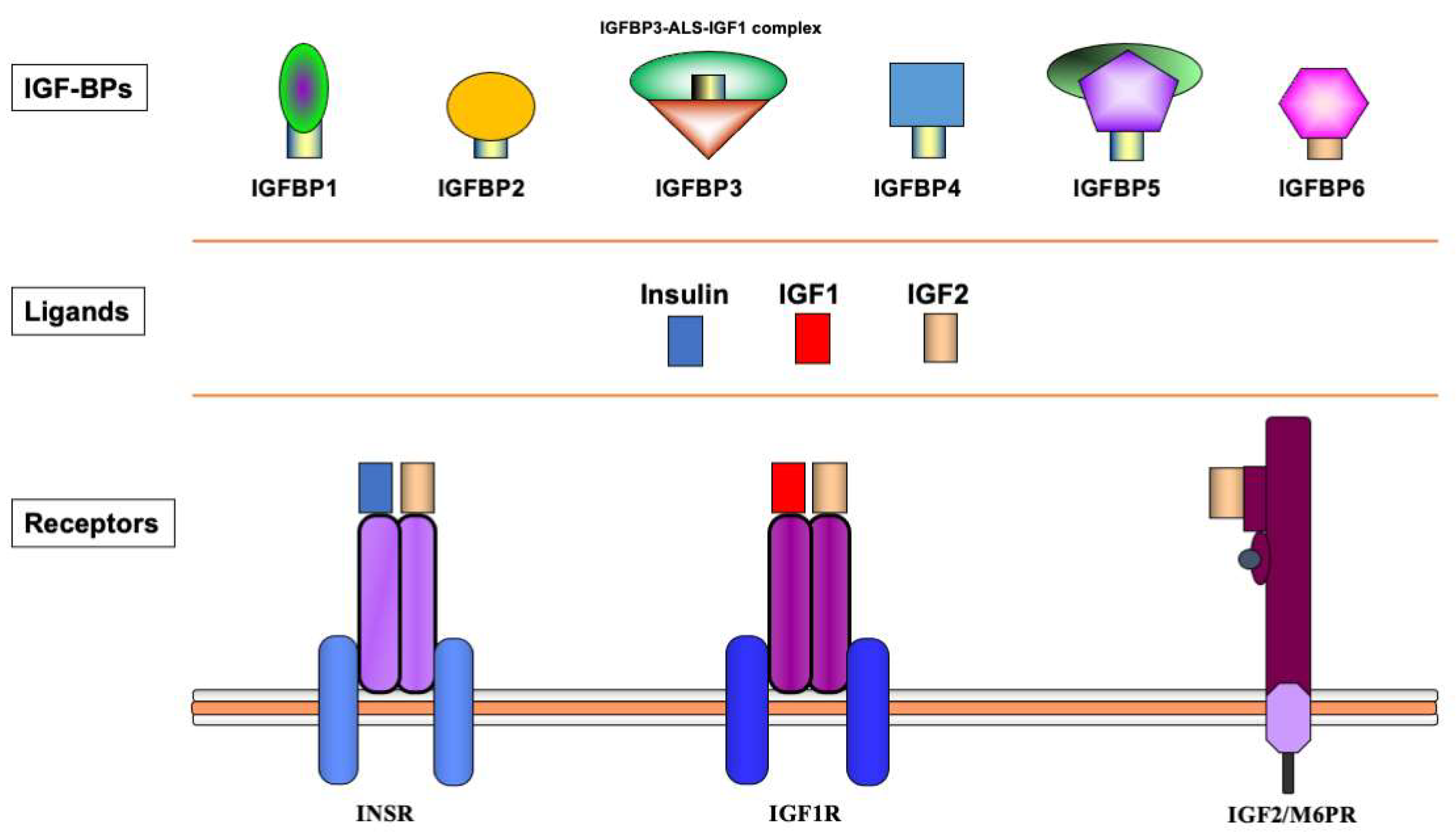
Figure 1. Schematic representation of the insulin-IGF network. The insulin-IGF family includes three ligands, three receptors and at least six IGFBPs. Experimental analyses provided evidence of cross-reactivity between ligands and receptors. Hence, IGF2 binds with high affinity to the InsR. In addition, both IGF1 and IGF2 activate hybrid receptors, formed by an IGF1R hemireceptor in association with an InsR hemireceptor (not shown). The IGF2/mannose 6-phosphate receptor is responsible for IGF2 recycling and is not involved in signaling. Most IGF1 in the circulation is bound to IGFBP3, which forms a multi-complex with an acid-labile subunit (ALS) and protects IGF1 from proteolytic degradation.
The Somatomedin Hypothesis was modified a number of times over the past decades, according to emerging scientific developments. It is widely accepted today that most, but not all, of the bioactivities of GH are mediated by liver-produced IGF1. While hepatic IGF1 biosynthesis is tightly dependent on GH stimulation, animal and human studies have identified a number of activities of IGF1 that are regarded as GH-independent.
The IGF network is recognized as a potential molecular target in cancer therapy and attempts are being made to translate preclinical results into medical protocols [23][24][25][26][27].
2. Insulin-like Growth Factor-1 (IGF1)
The IGF1 gene, located on chromosome band 12q22–q24, encompasses at least 90-kb of chromosomal DNA and contains six exons [28][29][30]. Alternative transcription initiation and splicing and various polyadenylation sequences result in a number of IGF1 mRNAs [31]. These mRNAs slightly differ in their coding sequences but mainly diverge in their 5′ and 3′ untranslated portions [32][33].
The IGF1 peptide (~7.65-kDa) consists of 70 amino acids, while IGF2 (~7.47-kDa) contains 67 amino acids [10][33]. IGF1 and IGF2 display a 62% homology in their amino acid sequences and there is a 40% similarity between both IGF1/2 and proinsulin [34][35]. Unlike insulin, wherein the connecting C-peptide is cleaved out during prohormone processing, the mature IGFs retain the C-domain that links the A and B domains. These structural divergences may explain the immunological distinction between IGFs and insulin that led to the historical discovery of non-suppressible insulin-like activity of IGF1. Furthermore, IGFs contain an extension to the A domain, termed the D domain, which is not present in insulin. Finally, both IGF precursors contain C-terminal E peptides. These peptides are cleaved during the processing of the prohormone.
Low IGF1 and high IG2 levels are detected during the prenatal period in rodents. Postnatal stages are associated with an increase in circulating IGF1 concentrations and the disappearance of IGF2 [31][36]. These early findings might have led to a mistaken interpretation of the roles of IGF2 and IGF1 as fetal and pubertal growth factors, respectively [37]. In humans, however, IGF2 and IGF1 are produced from prenatal to postnatal periods. In fact, endocrine IGF2 levels in adults are higher than IGF1. Of importance, liver-specific igf1 gene deletion in mice, while leading to a dramatic reduction in circulating IGF1 concentrations, had no major impact on body weight and length and femoral length. Hence, locally-produced (autocrine/paracrine) IGF1 seems to play a crucial role in organ and body growth and development [37][38].
In terms of the mechanisms that are responsible for regulation of the local production of IGF1, there is a marked variability between the different organs. In general, the biochemical machinery involved in IGF1 biosynthesis and action at the local level has been less well characterized than in the liver. Likewise, the paradigm that IGF1 production is tightly dependent on GH stimulation does not seem to apply to every tissue. Different tissue-specific hormones and growth factors have been shown to modulate IGF1 action. Thus, steroid hormones (e.g., androgens, estrogens) play a key role in IGF1 regulation in sex organs, while various neuropeptides control IGF1 activities in the brain.
The effect of polymorphisms in the IGF1 gene on endocrine IGF1 levels and cancer risk is variable [39]. IGF1 single-nucleotide polymorphisms (SNPs) individually account for up to 5% change in IGF1 concentrations, but no correlations have been observed between these polymorphisms and breast cancer risk. Hence, the impact of genetic variation in IGF1 on IGF1 levels does not appear to substantially modify breast cancer risk.
3. Insulin-like Growth Factor-1 Receptor (IGF1R)
IGF1 and IGF2 bind to and activate a shared, ubiquitously expressed, transmembrane receptor, the IGF1 receptor (IGF1R). IGF1R signals mitogenic, pro-survival and anti-apoptotic activities [12][18][40]. The IGF2/M6P receptor does not seem to participate in IGF signaling, and its main role is to target IGF2 for proteolytic degradation at the lysosome [15]. The IGF1R gene is located on the long arm of chromosome 15 (15q25–q26), and spans more than 100 kb of genomic DNA [41]. The gene encodes a 1367-amino acid pre-pro-receptor that is processed to yield mature α and β chains [32]. The mature receptor has an heterotetrameric structure that includes two extracellular α-subunits, involved in ligand binding, and two transmembrane β-subunits, containing a tyrosine kinase domain in their cytoplasmic portion [42]. As described below in more detail, the IGF1R is linked to various cytoplasmic second messenger molecules. The RAS-MAPK and PI3K signaling networks are the most important players in this context.
IGF1R action is fundamental for survival, as demonstrated by the lethality of mice in which the IGF1R gene was inactivated [43]. The IGF1R is abundantly expressed at every ontogenetic period, beginning from the oocyte stage [44][45]. At late fetal stages and during adulthood there is a marked decline in IGF1R mRNA concentrations [46]. This decrease is inversely correlated with the high proportion of terminally differentiated cells at these stages. The crucial role of IGF1R in organ growth and development is exemplified by the fact that IGF1R gene disruption results in animals that weigh 45% of their control littermates at the moment of birth. These animals display many developmental defects: hypoplasia, delayed bone development, defective skin formation, and atypical central nervous system morphology, etc. These animals die from respiratory collapse immediately after birth.
Further evidence for the role of IGF1R in development and growth is provided by the fact that chromosomal alterations involving the 15q26 locus (e.g., ring chromosome 15) are correlated with hemizygosity of the IGF1R locus and growth deficit [47]. Conversely, a patient with three copies of the IGF1R gene that resulted from duplication of the long arm of chromosome 15 had an height and weight above the 97th percentile. The patient exhibited an accelerated development [48]. These clinical studies highlight the link between IGF1R abundance and cell proliferation. Finally, analyses of multiple tumors showed high expression of IGF1R mRNA and protein. These tumors included breast, prostate, ovary, colon, hematopoietic, kidney, etc. These analyses led to the concept that IGF1R gene upregulation constitutes a common paradigm in cancer [49][50][51].
4. IGF Binding Proteins (IGFBPs)
Most of the IGF1 peptide in the blood is present in a ternary complex that includes, in addition to IGF1, a liver-produced glycoprotein (the acid-labile subunit, ALS) and an high-affinity-binding molecule, the IGFBP3 [20][52]. The proportion of free (or active) IGF1 is very low. At least six IGFBPs (IGFBP1–6) and a number of IGFBP-related molecules have been identified [53]. The predominant binding protein in blood is IGFBP3. Given its large molecular size, IGFBP3 cannot traverse the capillary membrane. The ternary complex formed by IGF1, IGFBP3 and ALS moderates IGF1 action by protecting the growth factor from proteolysis. As a result, IGF1’s half life is prolonged. In addition, as mentioned above, certain IGFBPs elicit their activities in an IGF-independent manner [22]. These findings are important in that they suggest that the spectrum of biological activities of IGFBPs goes beyond the characterized interactions with the IGF axis.
While the IGFBPs, in general, inhibit IGF actions, some IGFBPs display IGF-potentiating effects too [54][55]. IGFBP3 is regarded as an inhibitor of proliferation, eliciting a pro-apoptotic effect. A number of putative mechanisms have been postulated to explain IGFBP3 inhibitory activity. These mechanisms include sequestration of IGF1 from the receptor and binding competition with IGF1R [21]. In the specific case of prostate cancer, serum IGFBP2 was more than eight-fold higher in patients with metastatic disease compared to controls [56]. In contrast, a marked reduction in serum IGFBP3 was detected in patients with metastatic cancer. A significant correlation between serum IGFBP2 and prostate specific antigen (PSA) levels was observed, with a negative correlation between serum PSA and IGFBP3. These results suggest that IGFBPs participate in the growth regulation of prostate malignancy, and that variations in their blood levels may constitute biomarkers for prostate cancer.
A recent article by L. Bach provided an updated overview of IGFBPs [21]. IGFBPs control important biological processes such as proliferation, senescence, autophagy, migration, and angiogenesis. Furthermore, a number of mechanisms that are responsible for IGFBPs’ actions have been described, including modulation of other growth factors’ actions, transcriptional control, interaction with the sphingolipid pathway, binding to non-IGF molecules in the extracellular matrix, nuclear transport, etc. More studies are needed to evaluate the therapeutic potential of IGFBPs.
5. Signal Transduction
Ligand binding induces conformational changes that lead to autophosphorylation of the IGF1R β-subunit tyrosine kinase domain (comprising amino acids 973–1229) and subsequent ubiquitination of the receptor [57][58]. The IGF1R kinase domain contains an activation loop that includes three tyrosine residues (Tyr1,131, Tyr1,135 and Tyr1,136) that serve as autophosphorylation sites. Tyr1,135 and Tyr1,131 phosphorylation destabilizes the auto-inhibitory conformation of the activation loop, whereas Tyr1,136 phosphorylation stabilizes the catalytically optimized conformation. This step allows for substrate and ATP access [12][59]. Furthermore, the C-terminal domain includes a number of additional tyrosine and serine residues, such as Tyrs 1250, 1251 and 1316 and Sers 1280–1283. Phosphorylation of these sites is important in the context of IGF1R signaling. Mutation of all or some [60] of these residues affects the enzymatic activity as well as the biological properties of IGF1R [61][62]. The phosphorylated tyrosine residues serve as docking elements for other signaling molecules such as insulin receptor substrate (IRS)1-4 and Shc adaptor proteins. This event leads to activation of the PI3K/MAPK and the 14-3-3 pathways [18][63][64]. Of importance, constitutive phosphorylation of IGF1R constitutes a universal feature of all (or most) malignantly transformed cells.
In general, activation of the MAPK pathway leads to an increase in proliferation. On the other hand, activation of PI3K inhibits apoptosis and stimulates protein synthesis [65]. Phosphorylated IRS1 activates the 85-kDa regulatory subunit of PI3K, with ensuing activation of various downstream substrates, including AKT/PKB. In turn, AKT phosphorylation stimulates protein synthesis via mTOR activation, and elicits the anti-apoptotic effects of IGF1R via inactivation of BAD. In parallel, recruitment of Grb2/SOS by phosphorylated IRS1 or Shc leads to recruitment of Ras, with ensuing activation of the Ras-MAPK pathway. In addition to these networks, IGF1R signals also through the Janus kinase/signal transducer and activator of transcription pathway (JAK/STAT). Phosphorylation of JAK proteins leads to activation of STAT proteins. In particular, activation of STAT3 is critical for the potentially transforming activity of IGF1R. JNK kinases are also activated by IGF1R. IGF1 exerts inhibitory activities on JNK activation via phosphorylation and inhibition of MAP3K5/ASK1, which directly associates with IGF1R [66][67]. A simplified version of the signal transduction events mediated by IGF1R is shown in Figure 2.
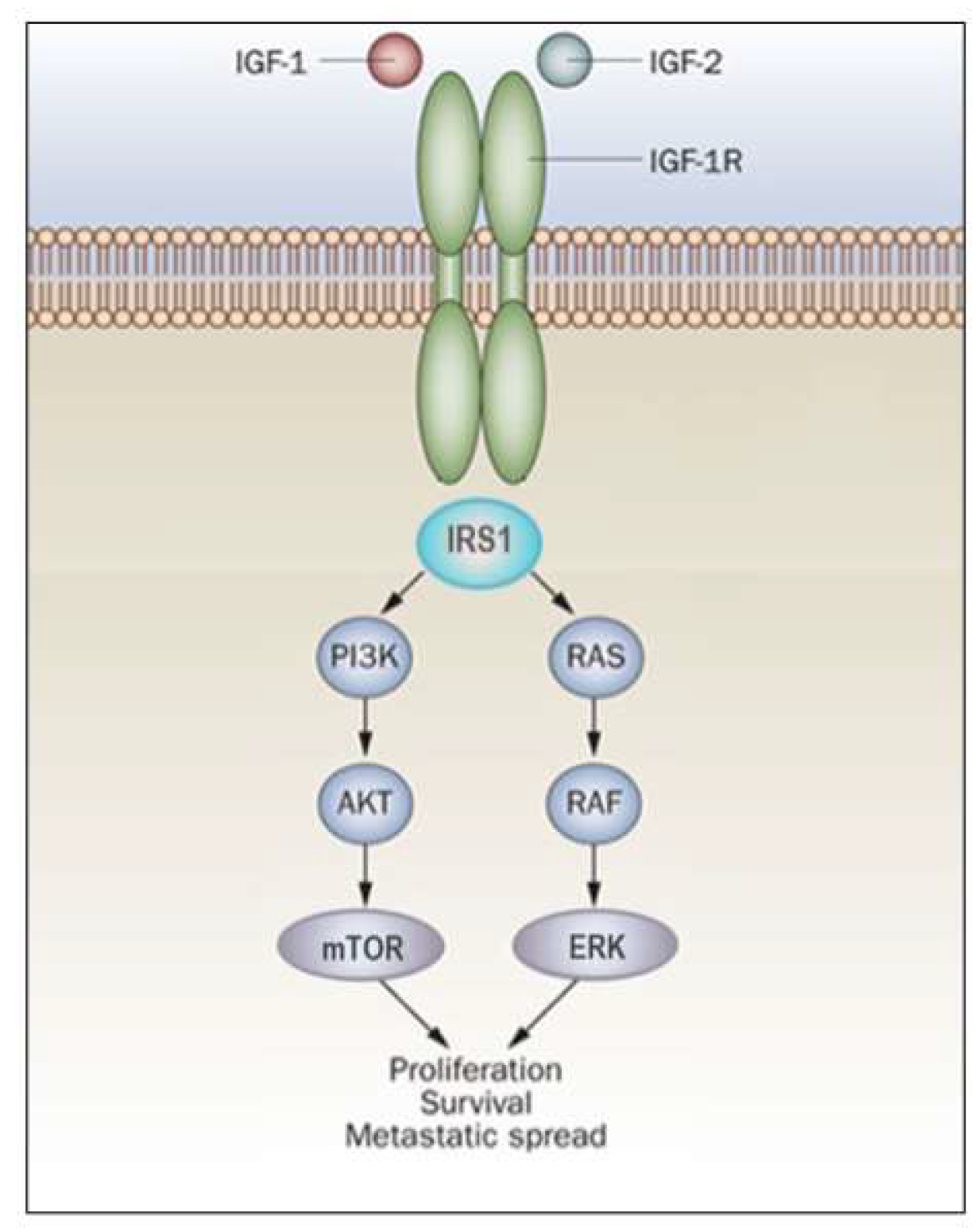
Figure 2. IGF1R signal transduction. The biological actions of IGF1 and IGF2 are transduced by the IGF1R and moderated by a family of at least six IGFBPs. Ligands bind with high affinity to the extracellular portion of IGF1R, and stimulate autophosphorylation of its tyrosine kinase (TK) domain. Upon activation of the IGF1R, IRSs become phosphorylated, with ensuing activation of two cascades, the RAS-MAP kinase (or ERK) and the PI3K-PDK1-Akt/PKB networks. The net consequence of the activation of these pathways is a boost in proliferation and a decrease in apoptosis.
Of major biological relevance, the substantial majority of the components of the signaling networks described above are shared by both IGF1R and InsR. This finding raises the question of how these receptors succeed in engaging in radically different biological activities despite a major overlap in their signaling molecules [14][68]. Several mechanisms were postulated to explain this paradox, including different distributions of InsR and IGF1R in tissues and organs, different subcellular distribution of the hormone-receptor complex, different internalization kinetics [69] and different hormone–receptor affinities [70][71][72]. In addition, various substrates and signaling mediators that are preferentially activated by either insulin or IGF1 have been identified. For example, the adapter protein Grb10 associates mainly with InsR, but not with IGF1R [73]. Likewise, the InsR, but not the IGF1R, interacts with pp120 [74]. Hence, differential activation of these and other substrates may explain, at least in part, the specificities of IGF1R and InsR.
6. Nuclear Import of IGF1R
Besides its typical mechanism of action at the cell-surface level, IGF1R is capable of translocating to the cell nucleus after modification by small ubiquitin-like modifier protein (SUMO)-1 [75][76][77][78]. Nuclear translocation is usually regarded as a ligand-dependent process, although some reports have provided evidence that mobilization of the cell-surface receptor may also take place in the absence of IGF1 stimulation. Nuclear IGF1R displays a number of activities that are classically correlated with transcription factors. These actions include DNA binding in a sequence-specific manner and transcription control [79][80]. Electrophoretic mobility shift assays in combination with super-shift assays using an IGF1R antibody allowed Sehat et al. to establish that IGF1R physically interacts with DNA [76]. The capacity of IGF1R to interact with DNA was investigated at a genome-wide level using chromatin immunoprecipitation assays. The majority (~80%) of IGF1R-enriched regions were intergenic (i.e., distal from annotated genes), whereas ~6% of these regions were present in introns and ~6% in exons. Data are in agreement with the idea that IGF1R binds to enhancer elements and functions as a transcription factor.
The finding that IGF1R migrates to the cell nucleus and interacts with DNA in a sequence-specific fashion supports the notion that in addition to the prototypic activities elicited by the receptor, IGF1R modulates biological processes at a genomic level [81]. Furthermore, while nuclear IGF1R’s presence was initially described in tumor cells (and, accordingly, inferred to constitute a pathologic type of localization), recent analyses have demonstrated that a pattern of nuclear IGF1R presence has also been seen in non-malignant human cells, including primary fibroblasts [82][83].
The potential consequences of nuclear IGF1R import in the clinic is a topic of great relevance [84]. While studies have shown that elevated nuclear IGF1R staining correlated with an adverse prognosis, the mechanisms responsible for nuclear IGF1R-mediated proliferation have not yet been investigated [85]. Furthermore, whilst inhibition of nuclear IGF1R migration by clathrin inhibitors correlated with major reductions in cell proliferation and invasiveness, the proteins that interact with IGF1R in the nucleus are yet to be identified [77][81]. Of major relevance, the discovery that MAPK, a key IGF1R cytoplasmic target, undergoes in itself nuclear translocation, raises questions regarding the mechanism/s of action of the IGF1R/MAPK intracellular signaling network [86][87][88]. In particular, it will be important to assess whether the nuclear transport of the cell-surface receptor and its cytoplasmic mediator take place in a coordinated fashion. Additional emerging questions include the following: (1) what is the functional significance of the joint nuclear localization of both IGF1R and MAPK?; and (2) does the pattern of IGF1R-MAPK nuclear migration reflect a generalized paradigm in cellular signaling?
7. Interaction of IGF1R with the p53 Genome Protection Axis
IGF1R expression is an important prerequisite for malignant transformation. Consistent with this concept, fibroblasts (termed R-) derived from igf1r knock-out mice are resistant to transformation by any of a series of oncogenes, including the SV40 large T antigen, activated ras, etc. [89][90]. However, introduction of a construct expressing a functional IGF1R into R- cells renders them sensitive to transformation. Nevertheless, IGF1R expression seems not to be an obligatory prerequisite, as suggested by the fact that various oncogenes induce transformation through alternative, IGF1R-independent pathways. In general, malignant cells exhibit augmented numbers of IGF1R on their cell surface as well as high levels of IGF1R mRNA. Constitutive activation (phosphorylation) of the receptor is regarded as a universal feature of cancer cells.
In addition to its mitogenic potential, the strong anti-apoptotic competence of IGF1R is undoubtedly the single most important trait that allows the receptor to play a key role in transformation. IGF1R protects cells from apoptotic death in multiple types of cultured cells as well as in vivo [91][92]. Increased concentrations of IGFIR at the cell-surface allowed cells to switch from a ‘non-mitogenic’ to a ‘mitogenic’ mode. Above a certain limit, cells acquired the ability to propagate in soft agar, a parameter of invasiveness [93][94]. It is relevant to question how terminally differentiated cells succeed in reducing IGF1R expression and, as a result, remain in a quiescent state. A potential mechanism that might be directly responsible for keeping IGF1R levels below a certain limit involves its transcriptional suppression by anti-oncogenes or tumor suppressors.
Tumor suppressor p53, the most frequently mutated molecule in human cancer, suppresses IGF1R promoter activity by ~90% as well as IGFIR mRNA levels [95]. In contradistinction, tumor-derived forms of p53 that result from mutations at codons 143, 248 or 273 enhanced promoter activity by ~2–4-fold [96]. The mechanism of action of p53 involves protein–protein interactions between p53 and components of the basal transcription machinery, including transcription factor Sp1. Extensive data indicate that the effects of p53 on cell cycle arrest are partly mediated by transcriptional suppression of the strongly anti-apoptotic IGF1R gene. Lack of inhibition of the IGF1R promoter by mutant p53 forms leads to a reduction in apoptosis, thus conferring an augmented survival capacity to cancer cells [97][98].
This entry is adapted from the peer-reviewed paper 10.3390/ijms241914882
References
- Salmon, W.D.; Daughaday, W.H. A hormonally controlled serum factor which stimulates sulfate incorporation by cartilage in vitro. J. Lab. Clin. Med. 1957, 49, 825–836.
- Salmon, W.D.J.; DuVall, M.R. In vitro stimulation of leucine incorporation into muscle and cartilage protein by a serum fraction with sulfation factor activity: Differentiation of effects from those of growth hormone and insulin. Endocrinology 1970, 87, 1168–1180.
- Daughaday, W.H.; Hall, K.; Raben, M.S.; Salmon, W.D.J.; van den Brande, J.L.; van Wyk, J.J. Somatomedin: Proposed designation for sulphation factor. Nature 1972, 235, 107.
- LeRoith, D.; Bondy, C.; Yakar, S.; Liu, J.-L.; Butler, A. The Somatomedin hypothesis: 2001. Endocrine Rev. 2001, 22, 53–74.
- Dulak, N.C.; Temin, H.M. Multiplication-stimulating activity for chicken embryo fibroblasts from rat liver cell conditioned medium: A family of small polypeptides. J. Cell Physiol. 1973, 81, 161–170.
- Rinderknecht, E.; Humbel, R.E. Polypeptides with nonsuppressible insulin-like and cell-growth promoting activities in human serum: Isolation, chemical characterization, and some biological properties of forms I and II. Proc. Natl. Acad. Sci. USA 1976, 73, 2365–2369.
- Froesch, E.R.; Buergi, H.; Ramseier, E.B.; Bally, P.; Labhart, A. Antibody-suppressible and nonsuppressible insulin-like activities in human serum and their physiologic significance. an insulin assay with adipose tissue of increased precision and specificity. J. Clin. Investig. 1963, 42, 1816–1834.
- Jakob, A.; Hauri, C.; Froesch, E.R. Nonsuppressible insulin-like activity in human serum. 3. Differentiation of two distinct molecules with nonsuppressible ILA. J. Clin. Investig. 1968, 47, 2678–2688.
- Daughaday, W.H.; Hall, K.; Salmon, W.D.J.; Van den Brande, J.L.; Van Wyk, J.J. On the nomenclature of the somatomedins and insulin-like growth factors. Mol. Endocrinol. 1987, 1, 1911–1912.
- Rinderknecht, E.; Humbel, R.E. The amino acid sequence of human insulin-like growth factor I and its structural homology with proinsulin. J. Biol. Chem. 1978, 253, 2769–2776.
- Miller, B.S.; Rogol, A.D.; Rosenfeld, R.G. The history of the insulin-like growth factor system. Horm. Res. Paed 2022, 95, 619–630.
- LeRoith, D.; Werner, H.; Beitner-Johnson, D.; Roberts, C.T., Jr. Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr. Rev. 1995, 16, 143–163.
- Rosenfeld, R.G. Insulin-like growth factors and the basis of growth. New Engl. J. Med. 2003, 349, 2184–2186.
- Werner, H.; Weinstein, D.; Bentov, I. Similarities and differences between insulin and IGF-I: Structures, receptors, and signaling pathways. Arch. Physiol. Biochem. 2008, 114, 17–22.
- MacDonald, R.G.; Pfeffer, S.R.; Coussens, L.; Tepper, M.A.; Brocklebank, C.M.; Mole, J.E.; Anderson, J.K.; Chen, E.; Czech, M.P.; Ullrich, A. A single receptor binds both insulin-like growth factor II and mannose-6-phosphate. Science 1988, 239, 1134–1137.
- Kiess, W. Molecular biology of the IGF-II/mannose-6-phosphate receptor. In The IGF System: Molecular Biology, Physiology, and Clinical Applications; Rosenfeld, R.G., Roberts, C.T., Jr., Eds.; Humana Press: Totowa, NJ, USA, 1999; pp. 89–109.
- Werner, H. Molecular biology of the Type 1 IGF receptor. In The IGF System: Molecular Biology, Physiology and Clinical Applications; Rosenfeld, R.G., Roberts, C.T., Jr., Eds.; Humana Press: Totowa, NJ, USA, 1999; pp. 63–88.
- Baserga, R.; Peruzzi, F.; Reiss, K. The IGF-1 receptor in cancer biology. Int. J. Cancer 2003, 107, 873–877.
- Bentov, I.; Werner, H. Insulin-like growth factor-I. In Handbook of Biologically Active Peptides; Kastin, A., Ed.; Elsevier Press: San Diego, CA, USA, 2006; pp. 1385–1392.
- Baxter, R.C. Insulin-like growth factor-binding proteins: Interactions with IGFs and intrinsic bioactivities. Am. J. Physiol. 2000, 278, 967–976.
- Bach, L. What happened to the IGF Binding Proteins? Endocrinology 2018, 159, 570–578.
- Oh, Y. IGF-independent regulation of breast cancer growth by IGF binding proteins. Breast Cancer Res. Treat. 1998, 47, 283–293.
- Crudden, C.; Girnita, A.; Girnita, L. Targeting the IGF-1R: The tale of the tortoise and the hare. Front. Endocrinol. 2015, 6, 64.
- Simpson, A.; Petnga, W.; Macaulay, V.M.; Weyer-Czernilofsky, U.; Bogenrieder, T. Insulin-like growth factor (IGF) pathway targeting in cancer: Role of the IGF axis and opportunities for future combination studies. Target. Oncol. 2017, 12, 571–597.
- Scotlandi, K.; Picci, P. Targeting insulin-like growth factor 1 receptor in sarcomas. Curr. Opin. Oncol. 2008, 20, 419–427.
- Sinai-Livne, T.; Pasmanik-Chor, M.; Cohen, Z.; Tsarfaty, I.; Werner, H.; Berger, R. Proteomic analysis of combined IGF1 receptor targeted therapy and chemotherapy identifies signatures associated with survival in breast cancer patients. Oncotarget 2020, 11, 1515–1530.
- Belfiore, A.; Frasca, F. IGF and insulin receptor signaling in breast cancer. J. Mammary Gland. Biol. Neoplasia 2008, 13, 381–406.
- Tricoli, J.V.; Rall, L.B.; Scott, J.; Bell, G.I.; Shows, T.B. Localization of insulin-like growth factor genes to human chromosomes 11 and 12. Nature 1984, 310, 784–786.
- Rotwein, P. Two insulin-like growth factor I messenger RNAs are expressed in human liver. Proc. Natl. Acad. Sci. USA 1986, 83, 77–81.
- Shimatsu, A.; Rotwein, P. Mosaic evolution of the insulin-like growth factors. Organization, sequence, and expression of the rat insulin-like growth factor I gene. J. Biol. Chem. 1987, 262, 7894–7900.
- Adamo, M.L.; Ben-Hur, H.; Roberts, C.T.J.; LeRoith, D. Regulation of start site usage in the leader exons of the rat insulin-like growth factor-I gene by development, fasting, and diabetes. Mol. Endocrinol. 1991, 5, 1677–1686.
- Werner, H.; Adamo, M.; Roberts, C.T., Jr.; LeRoith, D. Molecular and cellular aspects of insulin-like growth factor action. In Vitamins and Hormones; Litwack, G., Ed.; Academic Press: San Diego, CA, USA, 1994; Volume 48, pp. 1–58.
- Sussenbach, J.S. The gene structure of the insulin-like growth factor family. Prog. Growth Factor. Res. 1989, 1, 33–48.
- Blundell, T.L.; Bedarkar, S.; Humbel, R.E. Tertiary structures, receptor binding, and antigenicity of insulinlike growth factors. Fed. Proc. 1983, 42, 2592–2597.
- Daughaday, W.; Rotwein, P. Insulin-like growth factors I and II. Peptide, messenger ribonucleic acid and gene structures, serum, and tissue concentrations. Endocr. Rev. 1989, 10, 68–91.
- LeRoith, D.; Adamo, M.; Werner, H.; Roberts, C.T., Jr. Insulin-like growth factors and their receptors as growth regulators in normal physiology and pathological states. Trends Endocrinol. Metab. 1991, 2, 134–139.
- Yakar, S.; Adamo, M.L. Insulin-like growth factor 1 physiology: Lessons from mouse models. Endocrinol. Metab. Clin. N. Am. 2012, 41, 231–247.
- Yakar, S.; Liu, J.L.; Stannard, B.; Butler, A.; Accili, D.; Sauer, B.; LeRoith, D. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc. Natl. Acad. Sci. USA 1999, 96, 7324–7329.
- Patel, A.V.; Cheng, I.; Canzian, F.; Le Marchand, L.; Thun, M.J.; Berg, C.D.; Feigelson, H.S. IGF-1, IGFBP-1, and IGFBP-3 polymorphisms predict circulating IGF levels but not breast cancer risk: Findings from the Breast and Prostate Cancer Cohort Consortium (BPC3). PLoS ONE 2008, 3, e2578.
- Ullrich, A.; Gray, A.; Tam, A.W.; Yang-Feng, T.; Tsubowka, M.; Collins, C.; Henzel, W.; Lebon, T.; Kathuria, S.; Chen, F.; et al. Insulin-like growth factor I receptor primary structure: Comparison with insulin receptor suggests determinants that define functional specificity. EMBO J. 1986, 5, 2503–2512.
- Abbott, A.M.; Bueno, R.; Pedrini, M.T.; Murray, J.M.; Smith, R.J. Insulin-like growth factor I receptor gene structure. J. Biol. Chem. 1992, 267, 10759–10763.
- Frattali, A.L.; Pessin, J.E. Relationship between α subunit ligand occupancy and ß subunit autophosphorylation in insulin/insulin-like growth factor-I hybrid receptors. J. Biol. Chem. 1993, 268, 7393–7400.
- Baker, J.; Liu, J.-P.; Robertson, E.J.; Efstratiadis, A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell 1993, 75, 73–82.
- Bondy, C.A.; Werner, H.; Roberts, C.T., Jr.; LeRoith, D. Cellular pattern of insulin-like growth factor I (IGF-I) and type I IGF receptor gene expression in early organogenesis: Comparison with IGF-II gene expression. Mol. Endocrinol. 1990, 4, 1386–1398.
- Bondy, C.A.; Werner, H.; Roberts, C.T., Jr.; LeRoith, D. Cellular pattern of Type I insulin-like growth factor receptor gene expression during maturation of the rat brain: Comparison with insulin-like growth factors I and II. Neuroscience 1992, 46, 909–923.
- Werner, H.; Woloschak, M.; Adamo, M.; Shen-Orr, Z.; Roberts, C.T., Jr.; LeRoith, D. Developmental regulation of the rat insulin-like growth factor I receptor gene. Proc. Natl. Acad. Sci. USA 1989, 86, 7451–7455.
- Peoples, R.; Milatovich, A.; Francke, U. Hemizygosity at the insulin-like growth factor I receptor (IGF1R) locus and growth failure in the ring chromosome 15 syndrome. Cytogenet. Cell Genet. 1995, 70, 228–234.
- Okubo, Y.; Siddle, K.; Firth, H.; O’Rahilly, S.; Wilson, L.C.; Willatt, L.; Fukushima, T.; Takahashi, S.; Petry, C.J.; Saukkonen, T.; et al. Cell proliferation activities on skin fibroblasts from a short child with absence of one copy of the type 1 insulin-like growth factor receptor (IGF1R) gene and a tall child with three copies of the IGF1R gene. J. Clin. Endocrinol. Metab. 2003, 88, 5981–5988.
- Werner, H. The pathophysiological significance of IGF-I receptor overexpression: New insights. Ped. Endocrinol. Rev. 2009, 7, 2–5.
- Werner, H. Tumor suppressors govern insulin-like growth factor signaling pathways: Implications in metabolism and cancer. Oncogene 2012, 31, 2703–2714.
- Werner, H.; Bruchim, I. IGF-1 and BRCA1 signalling pathways in familial cancer. Lancet Oncol. 2012, 13, e537–e544.
- Domene, H.; Bengolea, S.V.; Martinez, A.S.; Ropelato, M.G.; Pennisi, P.; Scaglia, P.; Heinrich, J.; Jasper, H. Deficiency of the circulating IGF system associated with inactivation of the acid-labile subunit gene. New Engl. J. Med. 2004, 350, 570–577.
- Baxter, R.C.; Binoux, M.A.; Clemmons, D.R.; Conover, C.A.; Drop, S.L.S.; Holly, J.M.P.; Mohan, S.; Oh, Y.; Rosenfeld, R.G. Recomendations for nomenclature of the insulin-like growth factor binding protein superfamily. Endocrinology 1998, 139, 4036.
- Rechler, M.M. Insulin-like growth factor binding proteins. Vitam. Horm. 1993, 47, 1–114.
- Baxter, R.C. IGF binding proteins in cancer: Mechanistic and clinical insights. Nature Rev. Cancer 2014, 14, 329–341.
- Kanety, H.; Madjar, Y.; Dagan, Y.; Levi, J.; Papa, M.Z.; Pariente, C.; Goldwasser, B.; Karasik, A. Serum insulin-like growth factor-binding protein-2 (IGFBP-2) is increased and IGFBP-3 is decreased in patients with prostate cancer: Correlation with serum-specific antigen. J. Clin. Endocrinol. Metab. 1993, 77, 229–233.
- Girnita, L.; Girnita, A.; Larsson, O. Mdm2-dependent ubiquitination and degradation of the insulin-like growth factor-I receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 8247–8252.
- Vecchione, A.; Marchese, A.; Henry, P.; Rotin, D.; Morrione, A. The Grb10/Nedd4 complex regulates ligand-induced ubiquitination and stability of the insulin-like growth factor I receptor. Mol. Cell Biol. 2003, 23, 3363–3372.
- Favelyukis, S.; Till, J.H.; Hubbard, S.R.; Miller, W.T. Structure and autoregulation of the insulin-like growth factor 1 receptor kinase. Nat. Struct. Biol. 2001, 8, 1058–1063.
- Hernández-Sánchez, C.; Blakesley, V.; Kalebic, T.; Helman, L.; LeRoith, D. The role of the tyrosine kinase domain of the insulin-like growth factor-I receptor in intracellular signaling, cellular proliferation, and tumorigenesis. J. Biol. Chem. 1995, 270, 29176–29181.
- Gronborg, M.; Wulff, B.S.; Rasmussen, J.S.; Kjeldsen, T.; Gammeltoft, S. Structure-function relationship of the insulin-like growth factor-I receptor tyrosine kinase. J. Biol. Chem. 1993, 258, 23435–23440.
- Li, S.; Ferber, A.; Miura, M.; Baserga, R. Mitogenicity and transforming activity of the insulin-like growth factor-I receptor with mutations in the tyrosine kinase domain. J. Biol. Chem. 1994, 269, 32558–32564.
- Myers, M.G., Jr.; Sun, X.-J.; Cheatham, B.; Jachna, B.R.; Glasheen, E.M.; Backer, J.M.; White, M.F. IRS-1 is a common element in insulin and insulin-like growth factor-I signaling to the phosphatidylinositol 3’-kinase. Endocrinology 1993, 132, 1421–1430.
- Myers, M.G., Jr.; Backer, J.M.; Sun, X.-J.; Shoelson, S.E.; Hu, P.; Schlessinger, J.; Yoakim, M.; Schaffhausen, B.; White, M.F. IRS-1 activates the phosphatidylinositol 3’-kinase by associating with the src homology 2 domains of p85. Proc. Natl. Acad. Sci. USA 1992, 89, 10350–10354.
- Peruzzi, F.; Prisco, M.; Dews, M.; Salomoni, P.; Grassilli, E.; Romano, G.; Calabretta, B.; Baserga, R. Multiple signaling pathways of the insulin-like growth factor 1 receptor in protection from apoptosis. Mol. Cell Biol. 1999, 19, 7203–7215.
- Dupont, J.; Khan, J.; Qu, B.H.; Metzler, P.; Helman, L.; LeRoith, D. Insulin and IGF-1 induce different patterns of gene expression in mouse fibroblast NIH-3T3 cells: Identification by cDNA microarray analysis. Endocrinology 2001, 142, 4969–4975.
- Sepp-Lorenzino, L. Structure and function of the insulin-like growth factor I receptor. Breast Cancer Res. Treat. 1998, 47, 235–253.
- Dupont, J.; LeRoith, D. Insulin and insulin-like growth factor-I receptors: Similarities and differences in signal transduction. Horm. Res. 2001, 55 (Suppl. S2), 22–26.
- Zapf, A.; Hsu, D.; Olefsky, J.M. Comparison of the intracellular itineraries of insulin-like growth factor-I and insulin and their receptors in Rat-1 fibroblasts. Endocrinology 1994, 134, 2445–2452.
- De Meyts, P.; Whittaker, J. Structural biology of insulin and IGF1 receptors: Implications for drug design. Nature Rev. Drug Discov. 2002, 1, 769–783.
- De Meyts, P. Insulin/receptor binding: The last piece of the puzzle? What recent progress on the structure of the insulin/receptor complex tells us (or not) about negative cooperativity and activation. Bioessays 2015, 37, 389–397.
- Mastick, C.C.; Brady, M.J.; Printen, J.A.; Ribon, V.; Saltiel, A.R. Spatial determinants of specificity in insulin action. Mol. Cell. Biochem. 1998, 182, 65–71.
- Laviola, L.; Giorgino, F.; Chow, J.C.; Baquero, J.A.; Hansen, H.; Ooi, J.; Zhu, J.; Riedel, H.; Smith, R.J. The adapter protein Grb10 associates preferentially with the insulin receptor as compared with the IGF-I receptor in mouse fibroblasts. J. Biol. Chem. 1997, 99, 830–837.
- Najjar, S.M.; Blakesley, V.A.; Li Calzi, S.; Kato, H.; LeRoith, D.; Choice, C.V. Differential phosphorylation of pp120 by insulin and insulin-like growth factor I receptors: Role for the C-terminal domain of the beta-subunit. Biochemistry 1997, 36, 6827–6834.
- Sarfstein, R.; Pasmanik-Chor, M.; Yeheskel, A.; Edry, L.; Shomron, N.; Warman, N.; Wertheimer, E.; Maor, S.; Shochat, L.; Werner, H. Insulin-like growth factor-I receptor (IGF-IR) translocates to nucleus and autoregulates IGF-IR gene expression in breast cance cells. J. Biol. Chem. 2012, 287, 2766–2776.
- Sehat, B.; Tofigh, A.; Lin, Y.; Trocmé, E.; Liljedahl, U.; Lagergren, J.; Larsson, O. SUMOylation mediates the nuclear translocation and signaling of the IGF-1 receptor. Sci. Signal. 2010, 3, ra10.
- Aleksic, T.; Chitnis, M.M.; Perestenko, O.V.; Gao, S.; Thomas, P.H.; Turner, G.D.; Protheroe, A.S.; Howarth, M.; Macaulay, V.M. Type 1 insulin-like growth factor receptor translocates to the nucleus of human tumor cells. Cancer Res. 2010, 70, 6412–6419.
- Deng, H.; Lin, Y.; Badin, M.; Vasilcanu, D.; Strömberg, T.; Jernberg-Wiklund, H.; Sehat, B.; Larsson, O. Over-accumulation of nuclear IGF-1 receptor in tumor cells requires elevated expression of the receptor and the SUMO-conjugating enzyme Ubc9. Biochem. Biophys. Res. Commun. 2011, 404, 667–671.
- Aleksic, T.; Gray, N.E.; Wu, X.; Rieunier, G.; Osher, E.; Mills, J.; Verrill, C.; Bryant, R.J.; Han, C.; Hutchinson, K.; et al. Nuclear IGF-1R interacts with regulatory regions of chromatin to promote RNA polymerase II recruitment and gene expression associated with advanced tumor stage. Cancer Res. 2018, 78, 3497–3509.
- Warsito, D.; Sjöström, S.; Andersson, S.; Larsson, O.; Sehat, B. Nuclear IGF1R is a transcriptional co-activator of LEF1/TCF. EMBO Rep. 2012, 13, 244–250.
- Sarfstein, R.; Werner, H. Nuclear insulin and insulin-like growth factor-1 receptors: A novel paradigm in signal transduction. Endocrinology 2013, 154, 1672–1679.
- Solomon-Zemler, R.; Sarfstein, R.; Werner, H. Nuclear insulin-like growth factor-1 receptor (IGF1R) displays proliferative and regulatory activities in non-malignant cells. PLoS ONE 2017, 12, e0185164.
- Solomon-Zemler, R.; Pozniak, Y.; Geiger, T.; Werner, H. Identification of nucleolar protein NOM1 as a novel nuclear IGF1R-interacting protein. Mol. Genet. Metab. 2019, 126, 259–265.
- Codony-Servat, J.; Cuatrecasas, M.; Asensio, E.; Montironi, C.; Martínez-Cardús, A.; Marín-Aguilera, M.; Horndler, C.; Martínez-Balibrea, E.; Rubini, M.; Jares, P.; et al. Nuclear IGF-1R predicts chemotherapy and targeted therapy resistance in metastatic colorectal cancer. Br. J. Cancer 2017, 117, 1777–1786.
- Asmane, I.; Watkin, E.; Alberti, L.; Duc, A.; Marec-Berard, P.; Ray-Coquard, I.; Cassier, P.; Decouvelaere, A.V.; Ranchère, D.; Kurtz, J.E.; et al. Insulin-like growth factor type 1 receptor (IGF-1R) exclusive nuclear staining: A predictive biomarker for IGF-1R monoclonal antibody (Ab) therapy in sarcomas. Eur. J. Cancer 2012, 48, 3027–3035.
- Maik-Rachline, G.; Hacohen-Lev-Ran, A.; Seger, R. Nuclear ERK: Mechanism of translocation, substrates, and role in cancer. Int. J. Mol. Sci. 2019, 20, 1194.
- Flores, K.; Yadav, S.S.; Katz, A.A.; Seger, R. The nuclear translocation of mitogen-activated protein kinases: Molecular mechanisms and use as novel therapeutic target. Neuroendocrinology 2019, 108, 121–131.
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 2011, 1813, 1619–1633.
- Sell, C.; Rubini, M.; Rubin, R.; Liu, J.-P.; Efstratiadis, A.; Baserga, R. Simian virus 40 large tumor antigen is unable to transform mouse embryonic fibroblasts lacking type 1 insulin-like growth factor receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 11217–11221.
- Morrione, A.; DeAngelis, T.; Baserga, R. Failure of the bovine papillomavirus to transform mouse embryo fibroblasts with a targeted disruption of the insulin-like growth factor I receptor gene. J. Virol. 1995, 69, 5300–5303.
- Sell, C.; Baserga, R.; Rubin, R. Insulin-like growth factor I (IGF-I) and the IGF-I receptor prevent etoposide-induced apoptosis. Cancer Res. 1995, 55, 303–306.
- Resnicoff, M.; Abraham, D.; Yutanawiboonchai, W.; Rotman, H.L.; Kajstura, J.; Rubin, R.; Zoltick, P.; Baserga, R. The insulin-like growth factor I receptor protects tumor cells from apoptosis in vivo. Cancer Res. 1995, 55, 2463–2469.
- Resnicoff, M.; Burgaud, J.-L.; Rotman, H.L.; Abraham, D.; Baserga, R. Correlation between apoptosis, tumorigenesis, and levels of insulin-like growth factor I receptors. Cancer Res. 1995, 55, 3739–3741.
- Rubini, M.; Hongo, A.; D’Ambrosio, C.; Baserga, R. The IGF-I receptor in mitogenesis and transformation of mouse embryo cells: Role of receptor number. Exp. Cell Res. 1997, 230, 284–292.
- Werner, H.; Karnieli, E.; Rauscher, F.J., III; LeRoith, D. Wild type and mutant p53 differentially regulate transcription of the insulin-like growth factor I receptor gene. Proc. Nat. Acad. Sci. USA 1996, 93, 8318–8323.
- Ohlsson, C.; Kley, N.; Werner, H.; LeRoith, D. p53 regulates IGF-I receptor expression and IGF-I induced tyrosine phosphorylation in an osteosarcoma cell line: Interaction between p53 and Sp1. Endocrinology 1998, 139, 1101–1107.
- Werner, H.; Sarfstein, R.; LeRoith, D.; Bruchim, I. Insulin-like growth factor 1 signaling axis meets p53 genome protection pathways. Front. Oncol. 2016, 6, 159.
- Girnita, L.; Girnita, A.; Brodin, B.; Xie, Y.; Nilsson, G.; Dricu, A.; Lundeberg, J.; Wejde, J.; Bartolazzi, A.; Wiman, K.G.; et al. Increased expression of insulin-like growth factor I receptor in malignant cells expressing aberrant p53: Functional impact. Cancer Res. 2000, 60, 5278–5283.
This entry is offline, you can click here to edit this entry!