Colorectal cancer (CRC) is one of the most common and severe malignancies worldwide. Recent advances in diagnostic methods allow for more accurate identification and detection of several molecular biomarkers associated with this cancer. Classical prognostic genetic markers comprise mutations in several genes (e.g., APC, KRAS/BRAF, TGF-β, and TP53). Furthermore, CIN and MSI serve as chromosomal markers, while epigenetic markers include CIMP and many other candidates such as SERP, p14, p16, LINE-1, and RASSF1A. Results on the prognostic value of the most commonly used cell cycle-related markers in CRC demonstrated by immunohistochemical (IHC) methods in relation to patients' overall survival (OS) or disease-free survival (DFS), are inconsistent. However, it was possible to confirm such a role for cyclin B1, cyclin D1, proliferating cell nuclear antigen (PCNA) and Ki-67. The number of long non-coding RNAs (e.g., SNHG1, SNHG6, MALAT-1, CRNDE) and microRNAs (e.g., miR-20a, miR-21, miR-143, miR-145, miR-181a/b) associated with proliferation in CRC as confirmed prognostic markers is increasing. Despite the rather obvious limitations of IHC and new molecular techniques, the standardisation of methods for quantitative assessment of proliferation marker expression, or the understanding of endogenous and exogenous (environmental) mechanisms of accelerated cellular proliferation, requires further development.
- colorectal cancer
- proliferation markers
- immunohistochemistry
- modern biology molecular techniques
1. Introduction
Colorectal cancer (CRC) remains a major medical challenge worldwide, ranking third in prevalence and second among cancer-related death causes [1,2]. An increase can also be observed in CRC incidence in younger people (under 50 years of age), with those predisposed to CRC generally classified as ‘medium’ and ‘high’ risk groups [1,3,4].
The development of CRC is a multistage process. The numerous genetic alterations in CRC are reflected in morphological features that can be visualized by various molecular techniques [5,6]. A significant role in tumor initiation, growth, and metastasis is now attributed to cancer/tumor-initiating cells (CICs/TICs) or cancer stem cells (CSCs), which are capable of self-renewal and differentiation. Numerous studies support the ‘CSC hypothesis’, in which the essence of carcinogenesis is progressive colonic SC overpopulation. Research is ongoing into the biology of these cells, the identification of their molecular markers, and the mechanisms of CSC proliferation, differentiation, and resistance to treatment in CRC [7,8].
There are several theories regarding the sequence of events in the formation of CRC [9]. The first pathway of the ‘adenoma–carcinoma sequence’ describes a sequence of morphological alterations, from hyperplasia through dysplasia to the formation of malignant, invasive foci [5,10]. Pre-cancerous lesions, in this case, comprise adenomatous polyps [5], with the ‘adenoma–carcinoma sequence’ concept supplemented by early dysplastic lesions, known as aberrant crypt foci (ACF) [11,12]. The second theory of CRC formation, known as the mutator pathway, took its origin from the 1992 discovery of genetic alterations in patients with Lynch syndrome (LS), also known as hereditary non-polyposis CRC (HNPCC) [13].
Approximately 15% of CRC arises from genetic alterations. Several syndromes can be distinguished in the etiology of CRC, associated with a high lifetime risk of CRC due to the inheritance of mutations in a single gene. Specific ‘Mendelian’ CRC syndromes include familial adenomatous polyposis (FAP), with gene mutation of the adenomatous polyposis coli (APC) gene, LS genes (MSH2, MLH1, MSH6, PMS2), Peutz–Jeghers syndrome (LKB1/STK11), juvenile polyposis (SMAD4, BMPR1A), MUTYH-associated polyposis, and hereditary mixed polyposis (GREM1). All of these conditions, except for MUTYH-associated polyposis, are inherited in a dominant manner. However, there is a recessive version of HNPCC in which both copies of one of the DNA mismatch repair (MMRs) genes are mutated (reviewed in [14]).
A third theory of CRC development, the serrated pathway or hyperplastic polyp-carcinoma sequence, considers hyperplastic polyps (HPs) together with a subgroup of serrated polyps (SPs) as precursors of CRC [15]. It is now recognized that up to 10–30% of CRC cases arise through this alternative pathway, characterized by its genetic and epigenetic profile [16,17].
Together with changes in chromatin structure and DNA methylation, gene mutations in CRC lead to the dysregulation of signaling pathways responsible for cell proliferation, apoptosis, metabolism, differentiation, and survival [9,14,18].
The ’adenoma–carcinoma sequence’ is mainly characterized by a loss of proliferation control. In turn, in the serrated neoplasia pathway, a failure of apoptosis mechanisms is the most characteristic factor. However, asymmetric proliferation (shift of the zone of proliferation from the base to the lateral side) is typical of the architecturally distorted serrated crypt, a characteristic of sessile serrated lesions (SSLs) [15].
CRC is a typically malignant tumor characterized by genetic/epigenetic mutations in mutator genes (i.e., genes whose alterations accelerate mutations in other genes). However, the molecular and cellular alterations associated with the immortality (abnormal maintenance of proliferation) and autonomy of colorectal cells, as with other malignancies, remain unknown (reviewed in [19]). Studies also suggest that the mechanism linking abnormalities at the genetic (e.g., APC mutations) and cellular level (e.g., hyperplasia, dysplasia) between tumor initiation to metastasis is the excessive number of colonic CSCs. It also considers the symmetrical division of CSCs as an essential mechanism driving tumor growth, which may have therapeutic implications for patients with advanced CRC [20].
Due to the above, searching for optimal methods to evaluate tumor proliferation and for more sensitive markers with potential prognostic significance seems crucial. A prognostic factor is a variable that indicates the predicted natural course of the disease and can be used to estimate the chance of recovery or the likelihood of recurrence. Prognostic significance is particularly relevant to progression-free survival (PFS) and overall survival (OS). Prognostic factors are classified into tumor-related, host-related, and environmental [21,22].
2. Methods to Assess Cell Proliferation in Colorectal Cancer
In clinical practice and basic research, several methods exist for assessing the growth rate of normal and tumor cells. The most common is the assessment of (1) the “density” of ongoing mitoses in the tissue material, known as the mitotic index (the percentage of mitoses in the assessed pool of tumor cells per 1 mm2), the so-called mitotic rate, the rate at which cells enter the mitotic phase (M phase) (% of the cells/h) [23,24,25]; (2) the percentage of cells in the S phase by calculating the so-called bromo-, iododeoxyuridine labeling index (LIBrdIUdR) [26,27,28,29] with in vitro tritiated thymidine [30,31], or with a new thymidine analog, 5-ethynyl-2-deoxyuridine (EdU) labeling [32]; (3) IHC expression of classical proliferative markers (e.g., cyclins, proliferating cell nuclear antigen (PCNA), and Ki-67 [33,34,35,36,37]); (4) computed tomography (CT) with dual-layer spectral detector CT [38] or positron emission tomography (PET) [39,40,41].
The cancerogenic process of the colonic mucosa is associated with the development of cell proliferation abnormalities, which precede the onset of morphological alterations such as epithelial dysplasia. Individuals with gastrointestinal (GI) tract cancer risk factors and animals exposed to carcinogens mainly show an increase in the cell proliferation rate and abnormalities in the distribution of proliferating cells. The so-called extension of the proliferative compartment was observed even when the mucosa was not yet affected by morphological abnormalities. This proliferative feature seems to be related to the presence of defects in cell differentiation [42]. There is also a report in which a significantly lower expression of multi-gene proliferation signature (GPS) was observed in CRLMs, confirming lower levels of their proliferation using qRT-PCR and Ki-67 immunostaining. According to the authors, slow proliferation is a biological feature of both CRLMs and primary tumors with metastasis capacity [43].
In formalin-fixed, paraffin-embedded tissues, changes in DNA content or the expression of proteins involved in the cell cycle in dysplastic, precancerous, and neoplastic tissues of the human colon were most often comparatively assessed. However, changes in the expression of IHC markers (e.g., PCNA, p53, Ki-67) at different developmental stages of CRC were not always clear enough to serve as reliable prognostic markers [18,44,45].
2.1. Assessment of Mitosis in Cancer Tissues
The mitosis count/mitotic index in pathological samples allows for the assessment of tumor proliferative activity, facilitates tumor classification and diagnosis, assesses grade malignancy, determines aggressive behavior, allows for intratumoral lymphocyte counts, and may present prognostic significance [25,46]. The preferred sites for mitosis counting include invasive fronts (rich in viable tumor cells) or the periphery of the tumors. The tissue area for counting mitotic activity for different tumors was standardized as the number of mitoses in a fixed number of high-power fields (HPFs) (typically 10 fields of view at x 400 magnification) [46]. HPFs for digital pathology, different from glass-slide HPFs in conventional light microscopy, require re-evaluation [47]. The current recommendation for CRC is not to report the number of mitoses in HPFs, but to report them per square millimeter [25], or per 2 mm2 (this is approximately equivalent to 10 HPFs on modern microscopes) [24,46].
2.2. DNA Ploidy and Percentage of Cells in S Phase
Aneuploidy refers to an abnormal number of chromosomes in a cell, different from a multiplication of the haploid set, resulting from several genetic alterations. It reflects both gain/loss of whole chromosomes and unbalanced chromosome rearrangements (e.g., deletions, amplifications, translocations of large genome regions) [48]. For more than 100 years, aneuploidy has been postulated as a tumor-promoting factor, and its clinical relevance is still highlighted as a prognostic marker [49,50]. Interestingly, it has been suggested that tissue SCs have also developed their distinct response to aneuploidy, being able to survive and proliferate as aneuploid [49].
DNA content and ploidy were evaluated as prognostic factors in CRC [51,52,53,54], with DNA aneuploidy demonstrated to be a feature of tumors with a higher proliferation rate [52,54,55]. On the other hand, ploidy alone, determined by flow cytometry (FCM), had no prognostic significance in CRC (disease-free survival, DFS). In a group of more than 400 CRC patients, it was shown that nearly 73% of patients showed aneuploid tumors. Still, the DNA pattern was not correlated with either age, gender, location, differentiation, or stage of the tumors [56].
Review studies [57,58] and a meta-analysis [55] indicate a significant association of aneuploidy with tumor progression and a worse prognosis. An older meta-analysis (2007) showed that patients undergoing surgical resection of aneuploid CRC have a higher risk of death after five years [57]. Later meta-analysis (2015) including more than 7000 CRC patients showed a higher prevalence of aneuploidy in late versus early stage sporadic CRC (OD 1.51, 95% CI 1.37–1.67), indicating that genomic instability increases with CRC progression. In 54.1% of studies, a significant effect of aneuploidy on prognosis was described for OS, disease-specific survival (DSS), and recurrence (relapse)-free survival (RFS). Hence, aneuploidy may be considered as a tumor stage-specific prognostic marker [55].
Other methods to assess the proliferation of different cell populations in CRC include evaluating the number of cells in which DNA synthesis occurs using LIBrdIUdR, with tritiated thymidine [30,31] and EdU labeling [32]. Such procedures allow in vivo calculation of the S-phase fraction labeling index (LI), the duration of the S phase (Ts), and the potential tumor doubling time (Tpot) [59].
Evaluation of the binding index of BrdUrd/IdUrd/tritiated thymidine, etc., is possible (1) following the use of monoclonal antibodies (mAbs) against thymidine analogs in FCM or (2) by using the IHC method. Although these method variations are inexpensive and easy to perform, they are characterized by high subjectivity in the evaluation of specimens, poor reproducibility of results, and the lack of standardization between centers [31,52,56,60].
Studies from the 1990s showed that examining only the total and aneuploid LI in CRC is not sufficient as an indicator of proliferation, as Ts also can vary between tumors and even within a single tumor (from 4.0 to 28.6 h). The mean Tpot ranged from 1.7 to 21.4 days. None of the cellular kinetic parameters correlated with Dukes’ classification or histologic examination [61]. Wilson et al. showed that while IUdR assessed by FCM (IUdRfmc) and assessed by IHC (IUdRimm) correlated with each other, and their LIs were significantly higher in aneuploid than diploid tumors, no prognostic property of these markers was demonstrated [52]. Similar results were reported by other authors [54]. On the other hand, Palmqvist et al., using both IUdR detection techniques (FCM + IHC), demonstrated that patients with Dukes’ B tumors with higher IUdR LI (in invasive margin) and/or low Tpot (at both the invasive margin and the luminal border) had longer survival [27]. FCM studies on the prognostic value of the DNA index or S-phase fraction also did not demonstrate prognostic significance for disease recurrence in CRC stages II and III [53], survival in the overall group, or within stages [60]. In contrast, the kinetic parameters assessed by Michel et al. using in vivo injection of Brd and FCM, were independent prognostic factors in diploid tumors. These included lymph node (LN) involvement, ploidy, and Tpot in all tumors, and Tpot only in diploid tumors [59].
In summary, most studies failed to demonstrate the prognostic value of the CRC proliferation markers assessed. Moreover, using these methods, more accurate results for evaluating normal and tumor cell proliferation are obtained after analyzing material at different stages of CRC development. On the other hand, performing such tests before and during treatment allows one to predict the outcome of CRC radiotherapy. For example, BrdUrd LI before RT treatment of RC was not a predictor of early clinical and pathological tumor response. In contrast, the BrdUrd LI ratio before/after RT was correlated with the inhibition of proliferation in responsive tumors. Thus, the rapid growth rate of preoperatively irradiated rectal cancer was a favorable prognostic factor [62].
2.3. Immunohistochemical Methods for the Detection of Proliferative Markers
The immunohistochemical (IHC) technique is based on antibodies against specific antigens in tissues and cells. In histopathology, IHC testing is most commonly performed on formalin-fixed, paraffin-embedded tissues that can be stored for long periods of time [63,64]. Increasingly, tissue microarrays (TMAs), which contain selected tissue material from tumors, normal tissues (control), and tumor metastases on a single slide, are being used for IHC. Although the cost of producing TMAs remains high, their selection saves labor time and the number of reagents used (including sometimes expensive antibodies), allowing for better result reproducibility [65]. The IHC markers most commonly used to assess tumor proliferation rate (including CRC) include thymidylate synthase (TS), cyclins, proliferating cell nuclear antygen (PCNA) and Ki-67 antigen. The IHC expression of a given proliferation marker has most often been related to OS and DFS in CRC patients. Meta-analyses have been able to confirm such a role for overexpression of cyclin B1 (better 5-year survival) [66], overexpression of cyclin D1 (poor OS and DFS) [67,68], increased expression of PCNA (poor OS and cancer-specific survival, CSS) [69] and overexpression of Ki-67 (poor OS) [70].
2.4. Modern Molecular Biology Techniques for the Assessment of Proliferative Markers in CRC
With the rapid development of complex molecular biology techniques (e.g., qRT-PCR, in situ hybridization (ISH), RNA/DNA sequencing, NGS, and DNA methylation detection methods), there is a constant search for new biomarkers of cellular proliferation with potential diagnostic, prognostic, and/or predictive significance in cancers including CRC [43,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88].
Quantitative RT-PCR is generally used as the ‘gold standard’ method to measure RNA expression [43,72,77,86,87]. In situ hybridization is a research tool to detect protein production and provides invaluable information regarding the localization of gene expression in heterogeneous tissues. For example, it was used to detect Ki-67 mRNA in CRC tissues with the digoxigenin-labelled cRNA probe [71].
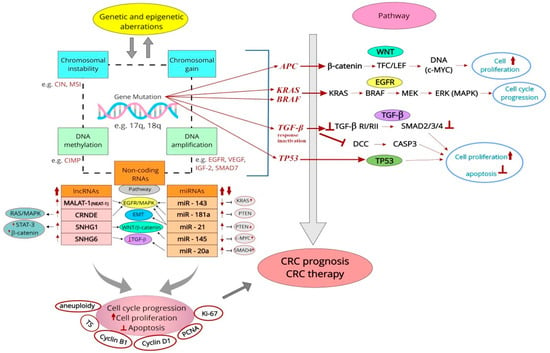
RNA sequencing is used to study the expression of non-coding RNAs (ncRNAs) [85,86], often complementary to methods for assessing protein expression (e.g., IHC, BrdU staining, Western blotting, qRT-PCR, and ISH). Among the sequencing techniques, NGS is currently the only method that enables the parallel sequencing of thousands of short DNA sequences in a single assay, replacing many less advanced profiling technologies. NGS is used to analyze the genome (whole and partial genome), methylome, transcriptome, or available chromatin using techniques including DNA-Seq, RNA-Seq, or chromatin profiling with methods such as ChIP-Seq. This technology offers a better approach for detecting multiple genetic changes with a minimal amount of DNA. What is particularly important is that it is also possible to sequence RNA transcripts from single cells (scRNA-Seq) [89].
Detection methods for DNA methylation in CRC include methylation-specific polymerase chain reaction (MSR), DNA sequencing (e.g., bisulfide sequencing, pyrosequencing), methylation-specific high resolution melting curve analysis (MS-HRM), and MethyLight assay (reviewed in [88]).
2.5. Positron Emission Tomography (PET) to Assess Tumor Growth Rate
PET is the most specific and sensitive method of in vivo molecular interaction and pathway imaging, finding an increasing number of applications in oncology [90]. This non-invasive technique for the functional imaging and assessment of CRC growth rate is based on the use of labelled 18-fluoro-3-deoxy-3-fluorothymidine (FLT). The method can reveal the spatial organization of proliferating cells in the tumor and allows for multiple simultaneous in vivo measurements. However, there are some correlations between FLT uptake and tumor proliferative activity [39,40]. FLT was reported to have high sensitivity in detecting extrahepatic disease but poor sensitivity in imaging CRC liver metastases [40]. A better and currently the most commonly used tracer in CRC is 18F-labelled 2-fluoro-2-deoxy-D-glucose (18F-FDG), with its usefulness resulting from increased glucose consumption by malignant cells. Therefore, this tracer’s uptake is closely linked to cancer cell proliferation, which depends mainly on glycolysis for energy. Many signal transduction pathways in the malignant transformation of cancer cells are regulated by glycolytic metabolism [91]. Therefore, the combination of PET and 18F-FDG has become an established tool for diagnostic tumor imaging and complete preoperative staging in CRC [40,41,92]. PET–18F-FDG results may have implications for the therapeutic management of patients with CRC [92,93] including metastatic CRC [41]. One review recognized that PET in CRC also allows for the metabolic characterization of lesions suspected of recurrence or the identification of latent metastatic disease [92]. Comparative studies indicate lower FLT versus FDG uptake in patients with CRC. However, no correlation was shown between the two radiotracers used and the proliferative activity assessed by the Ki-67 index [94]. A later meta-analysis only confirmed a moderate correlation between 18F-FDG uptake and Ki-67 expression in CRC [95]. A recent study by Watanabe et al. indicated that tumor proliferation in CRLM is reflected by the standardized uptake value (SUV) from FDG-PET. In addition, the authors showed a high correlation between SUV and Ki-67 expression. SUV was also shown to include factors of glucose metabolism (expression of hypoxia-inducible factor 1 alpha (HIF-1α), pyruvate kinase M2 (PKM2), and glucose transporter 1 (GLUT1)). Thus, this test can be a valuable method to assess the proliferative and metabolic viability of the tumor in advanced CRLM [41].
The remaining limitations of PET comprise its high cost and the lack of necessary equipment in cancer centers, limiting the potential for multidisciplinary PET studies. The most significant limitation for the patient is the need for the administration of radioactive tracers, resulting in potential radiation exposure [90].
3. References
- Ahmed, M. Colon Cancer: A Clinician's Perspective in 2019. Gastroenterology Res. 2020, 13, 1-10. doi: 10.14740/gr1239.
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17-48. doi: 10.3322/caac.21763.
- Morgan, E.; Arnold, M.; Gini, A.; Lorenzoni, V.; Cabasag, C.J.; Laversanne, M.; Vignat, J.; Ferlay, J.; Murphy, N.; Bray, F. Global burden of colorectal cancer in 2020 and 2040: incidence and mortality estimates from GLOBOCAN. Gut. 2023, 72, 338-344. doi: 10.1136/gutjnl-2022-327736.
- Xi, Y.; Xu, P. Global colorectal cancer burden in 2020 and projections to 2040. Oncol. 2021, 14, 101174. doi: 10.1016/j.tranon.2021.101174.
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. Engl. J. Med. 1988, 319, 525-532. doi: 10.1056/NEJM198809013190901.
- Dariya, B.; Aliya, S.; Merchant, N.; Alam, A.; Nagaraju, G.P. Colorectal Cancer Biology, Diagnosis, and Therapeutic Approaches. Rev. Oncog. 2020, 25, 71-94. doi: 10.1615/CritRevOncog.2020035067.
- Dieter, SM, Ball, CR, Hoffmann, CM, Nowrouzi, A, Herbst, F, Zavidij, O, Abel, U, Arens, A, Weichert, W, Brand, K, et al. Distinct types of tumor-initiating cells form human colon cancer tumors and metastases. Cell Stem Cell. 2011, 9, 357-365. doi: 10.1016/j.stem.2011.08.010. PMID: 21982235.
- Dieter, S.M.; Glimm, H.; Ball, C.R. Colorectal cancer-initiating cells caught in the act. EMBO Mol. Med. 2017, 9, 856-858. doi: 10.15252/emmm.201707858.
- Bosman, F.; Yan, P. Molecular pathology of colorectal cancer. J. Pathol. 2014, 65, 257-266. doi: 10.5114/pjp.2014.48094.
- Kinzler, K.W.; Vogelstein, B. Lessons from hereditary colorectal cancer. 1996, 87, 159-170. doi: 10.1016/s0092-8674(00)81333-1.
- Cheng, L.; Lai, M.D. Aberrant crypt foci as microscopic precursors of colorectal cancer. World J. Gastroenterol. 2003, 9, 2642-2649. doi: 10.3748/wjg.v9.i12.2642.
- Kowalczyk, M.; Orłowski, M.; Klepacki, Ł.; Zinkiewicz, K.; Kurpiewski, W.; Kaczerska, D.; Pesta, W.; Zieliński, E.; Siermontowski, P. Rectal aberrant crypt foci (ACF) as a predictor of benign and malignant neoplastic lesions in the large intestine. BMC Cancer. 2020, 20, 133. doi: 10.1186/s12885-020-6590-4.
- Lynch, H.T.; Kimberling, W.; Albano, W.A.; Lynch, J.F.; Biscone, K.; Schuelke, G.S.; Sandberg, A.A.; Lipkin, M.; Deschner, E.E.; Mikol, Y.B.; et al. Hereditary nonpolyposis colorectal cancer (Lynch syndromes I and II). I. Clinical description of resource. Cancer. 1985, 56, 934-938. doi: 10.1002/1097-0142(19850815)56:4<934::aid-cncr2820560439>3.0.co;2-i.
- Fearon, E.R. Molecular genetics of colorectal cancer. Annu Rev. Pathol. 2011, 6, 479-507. doi: 10.1146/annurev-pathol-011110-130235.
- Kim, J.H.; Kang, G.H. Evolving pathologic concepts of serrated lesions of the colorectum. Pathol. Transl. Med. 2020, 54, 276-289. doi: 10.4132/jptm.2020.04.15.
- Patai, A.V.; Molnár, B.; Tulassay, Z.; Sipos, F. Serrated pathway: alternative route to colorectal cancer. World J. Gastroenterol. 2013, 19, 607-615. doi: 10.3748/wjg.v19.i5.607.
- Thorlacius, H.; Takeuchi, Y.; Kanesaka, T.; Ljungberg, O.; Uedo, N.; Toth, E. Serrated polyps - a concealed but prevalent precursor of colorectal cancer. J. Gastroenterol. 2017, 52, 654-661. doi: 10.1080/00365521.2017.1298154.
- Polyak, K.; Hamilton, S.R.; Vogelstein, B.; Kinzler, K.W. Early alteration of cell-cycle-regulated gene expression in colorectal neoplasia. J. Pathol. 1996, 149, 381-387. PMID: 8701978; PMCID: PMC1865297.
- Zhu, S.; Wang, J.; Zellmer, L.; Xu, N.; Liu, M.; Hu, Y.; Ma, H.; Deng, F.; Yang, W.; Liao, D.J. Mutation or not, what directly establishes a neoplastic state, namely cellular immortality and autonomy, still remains unknown and should be prioritized in our research. Cancer. 2022, 13, 2810-2843. doi: 10.7150/jca.72628.
- Boman, B.M.; Huang, E. Human colon cancer stem cells: a new paradigm in gastrointestinal oncology. Clin. Oncol. 2008, 26, 2828-2838. doi: 10.1200/JCO.2008.17.6941.
- Simms, L.; Barraclough, H.; Govindan, R. Biostatistics primer: what a clinician ought to know--prognostic and predictive factors. Thorac. Oncol. 2013, 8, 808-813. doi: 10.1097/JTO.0b013e318292bdcd.
- National Cancer Institute Dictionary of Cancer Terms. Definition of prognostic factor. Available at: http://www.cancer.gov/dictionary?CdrID=44245. Accessed May 14, 2023.
- Oshima, C.T.; Iriya, K.; Forones, N.M. Ki-67 as a prognostic marker in colorectal cancer but not in gastric cancer. Neoplasma. 2005, 52, 420-4. PMID: 16151588.
- Ahadi, M.; Sokolova, A.; Brown, I.; Chou, A.; Gill, A.J. The 2019 World Health Organization Classification of appendiceal, colorectal and anal canal tumours: an update and critical assessment. Pathology. 2021, 53, 454-461. doi: 10.1016/j.pathol.2020.10.010.
- Cree, I.A.; Tan, P.H.; Travis, W.D.; Wesseling, P.; Yagi, Y.; White, V.A.; Lokuhetty, D.; Scolyer, R.A. Counting mitoses: SI(ze) matters! Pathol. 2021, 34, 1651-1657. doi: 10.1038/s41379-021-00825-7.
- Potten, C.S.; Kellett, M.; Roberts, S.A.; Rew, D.A.; Wilson, G.D. Measurement of in vivo proliferation in human colorectal mucosa using bromodeoxyuridine. Gut. 1992, 33, 71-78. doi: 10.1136/gut.33.1.71.
- Palmqvist, R.; Oberg, A.; Bergström, C.; Rutegård, J.N.; Zackrisson, B.; Stenling, R. Systematic heterogeneity and prognostic significance of cell proliferation in colorectal cancer. J. Cancer. 1998, 77, 917-925. doi: 10.1038/bjc.1998.152.
- Salud, A.; Porcel, J.M.; Raikundalia, B.; Camplejohn, R.S.; Taub, N.A. Prognostic significance of DNA ploidy, S-phase fraction, and P-glycoprotein expression in colorectal cancer. Surg. Oncol. 1999, 72, 167-174. doi: 10.1002/(sici)1096-9098(199911)72:3<167::aid-jso10>3.0.co;2-h.
- Rew, D.A.; Wilson, G.D. Cell production rates in human tissues and tumours and their significance. Part 1: an introduction to the techniques of measurement and their limitations. J. Surg. Oncol. 2000, 26, 227-238. doi: 10.1053/ejso.1999.0781.
- Biasco, G.; Paganelli, G.M.; Santucci, R.; Brandi, G.; Barbara, L. Methodological problems in the use of rectal cell proliferation as a biomarker of colorectal cancer risk. Cell Biochem. Suppl. 1994, 19, 55-60. PMID: 7823606.
- Paganelli, G.M.; Lalli, E.; Facchini, A.; Biasco, G.; Santucci, R.; Brandi, G.; Barbara, L. Flow cytometry and in vitro tritiated thymidine labeling in normal rectal mucosa of patients at high risk of colorectal cancer. J. Gastroenterol. 1994, 89, 220-224. PMID: 8304307.
- Alowaidi, F.; Hashimi, S.M.; Alqurashi, N.; Alhulais, R.; Ivanovski, S.; Bellette, B.; Meedenyia, A.; Lam, A.; Wood, S. Assessing stemness and proliferation properties of the newly established colon cancer 'stem' cell line, CSC480 and novel approaches to identify dormant cancer cells. Rep. 2018, 39, 2881-2891. doi: 10.3892/or.2018.6392.
- Gerdes, J.; Lemke, H.; Baisch, H.; Wacker, H.H.; Schwab, U.; Stein, H. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. Immunol. 1984, 133, 1710-1715. PMID: 6206131.
- Scholzen, T.; Gerdes, J. The Ki-67 protein: from the known and the unknown. Cell Physiol. 2000, 182, 311-322. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9.
- Hall, P.A.; Levison, D.A. Review: assessment of cell proliferation in histological material. Clin. Pathol. 1990, 43, 184-192. doi: 10.1136/jcp.43.3.184.
- Sawtell, R.M.; Rew, D.A.; Stradling, R.N.; Wilson, G.D. Pan cycle expression of proliferating cell nuclear antigen in human colorectal cancer and its proliferative correlations. Cytometry. 1995, 22, 190-199. doi: 10.1002/cyto.990220306.
- Sobecki, M.; Mrouj, K.; Colinge, J.; Gerbe, F.; Jay, P.; Krasinska, L.; Dulic, V.; Fisher, D. Cell-Cycle Regulation Accounts for Variability in Ki-67 Expression Levels. Cancer Res. 2017, 77, 2722-2734. doi: 10.1158/0008-5472.CAN-16-0707.
- Wang, Y.L.; Zhang, H.W.; Mo, Y.Q.; Zhong, H.; Liu, W.M.; Lei, Y.; Lin, F. Application of dual-layer spectral detector computed tomography to evaluate the expression of Ki-67 in colorectal cancer. Chin. Med. Assoc. 2022, 85, 610-616. doi: 10.1097/JCMA.0000000000000706. Epub 2022 May 2. PMID: 35286294.
- Francis, D.L.; Freeman, A.; Visvikis, D.; Costa, D.C.; Luthra, S.K.; Novelli, M.; Taylor, I.; Ell, P.J. In vivo imaging of cellular proliferation in colorectal cancer using positron emission tomography. Gut. 2003, 52, 1602-1606. doi: 10.1136/gut.52.11.1602.
- Francis, D.L.; Visvikis, D.; Costa, D.C.; Arulampalam, T.H.; Townsend, C.; Luthra, S.K.; Taylor, I.; Ell, P.J. Potential impact of [18F]3'-deoxy-3'-fluorothymidine versus [18F]fluoro-2-deoxy-D-glucose in positron emission tomography for colorectal cancer. J. Nucl. Med. Mol. Imaging. 2003, 30, 988-994. doi: 10.1007/s00259-003-1187-0.
- Watanabe, A.; Harimoto, N.; Yokobori, T.; Araki, K.; Kubo, N.; Igarashi, T.; Tsukagoshi, M.; Ishii, N.; Yamanaka, T.; Handa, T.; et al. FDG-PET reflects tumor viability on SUV in colorectal cancer liver metastasis. J. Clin. Oncol. 2020, 25, 322-329. doi: 10.1007/s10147-019-01557-0.
- Biasco, G.; Paganelli, G.M.; Miglioli, M.; Barbara, L. Cell proliferation biomarkers in the gastrointestinal tract. Cell Biochem Suppl. 1992, 16G, 73-78. doi: 10.1002/jcb.240501114.
- Anjomshoaa, A.; Nasri, S.; Humar, B.; McCall, J.L.; Chatterjee, A.; Yoon, H.S.; McNoe, L.; Black, M.A.; Reeve, A.E. Slow proliferation as a biological feature of colorectal cancer metastasis. J. Cancer. 2009, 101, 822-828. doi: 10.1038/sj.bjc.6605229.
- Tomita, T. DNA ploidy and proliferating cell nuclear antigen in colonic adenomas and adenocarcinomas. Dis. Sci. 1995, 40, 996-1004. doi: 10.1007/BF02064188.
- Barletta, A.; Marzullo, F.; Pellecchia, A.; Montemurro, S.; Labriola, A.; Lomonaco, R.; Grammatica, L.; Paradiso, A. DNA flow cytometry, p53 levels and proliferative cell nuclear antigen in human colon dysplastic, precancerous and cancerous tissues. Anticancer Res. 1998, 18, 1677-1682. PMID: 9673389.
- Yigit, N.; Gunal, A.; Kucukodaci, Z.; Karslioglu, Y.; Onguru, O.; Ozcan, A. Are we counting mitoses correctly? Diagn. Pathol. 2013, 17, 536-539. doi: 10.1016/j.anndiagpath.2013.05.005. Epub 2013 Jun 24. PMID: 23806202.
- Kim, D.; Pantanowitz, L.; Schüffler, P.; Yarlagadda, D.V.K.; Ardon, O.; Reuter, V.E.; Hameed, M.; Klimstra, D.S.; Hanna, M.G. (Re) Defining the High-Power Field for Digital Pathology. Pathol. Inform. 2020, 11, 33. doi: 10.4103/jpi.jpi_48_20.
- Orr, B.; Godek, K.M.; Compton, D. Aneuploidy. Biol. 2015, 25, R538-542. doi: 10.1016/j.cub.2015.05.010.
- Brás, R.; Sunkel, C.E.; Resende, L.P. Tissue stem cells: the new actors in the aneuploidy field. Cell Cycle. 2019, 18, 1813-1823. doi: 10.1080/15384101.2019.1635867.
- Ben-David, U.; Amon, A. Context is everything: aneuploidy in cancer. Rev. Genet. 2020, 21, 44-62. doi: 10.1038/s41576-019-0171-x.
- Lazaris, A.C.; Davaris, P.; Nakopoulou, L.; Theodoropoulos, G.E.; Koullias, G.; Golematis, B.C. Correlation between immunohistochemical expression of proliferating cell nuclear antigen and flow cytometry parameters in colorectal neoplasia. Colon Rectum. 1994, 37, 1083-1089. doi: 10.1007/BF02049808.
- Wilson, M.S.; Anderson, E.; Bell, J.C.; Pearson, J.M.; Haboubi, N.Y.; James, R.D.; Schofield, P.F. An evaluation of five different methods for estimating proliferation in human colorectal adenocarcinomas. Oncol. 1994, 3, 263-273. doi: 10.1016/0960-7404(94)90028-0.
- Chen, Y.T.; Henk, M.J.; Carney, K.J.; Wong, W.D.; Rothenberger, D.A.; Zheng, T.; Feygin, M.; Madoff, R.D. Prognostic significance of tumor markers in colorectal cancer patients: DNA index, S-phase fraction, p53 expression, and Ki-67 index. Gastrointest. Surg. 1997, 1, 266-272; discussion 273. doi: 10.1016/s1091-255x(97)80119-9.
- Le Pessot, F.; Michel, P.; Paresy, M.; Lemoine, F.; Hellot, M.F.; Paillot, B.; Scotte, M.; Peillon, C.; Hemet, J. Cell proliferation in colorectal adenocarcinomas: comparison between Ki-67 immunostaining and bromodeoxyuridine uptake detected by immunohistochemistry and flow cytometry. Res. Pract. 2001, 197, 411-418. doi: 10.1078/0344-0338-00054.
- Laubert, T.; Freitag-Wolf, S.; Linnebacher, M.; König, A.; Vollmar, B.; Habermann, J.K. North German Tumorbank of Colorectal Cancer (ColoNet) consortium. Stage-specific frequency and prognostic significance of aneuploidy in patients with sporadic colorectal cancer--a meta-analysis and current overview. J. Colorectal Dis. 2015, 30, 1015-1028. doi: 10.1007/s00384-015-2259-x.
- Lin, J.K.; Chang, S.C.; Yang, S.H.; Jiang, J.K.; Chen, W.C.; Lin, T.C. Prognostic value of DNA ploidy patterns of colorectal adenocarcinoma. Hepatogastroenterology. 2003, 50, 1927-1932. PMID: 14696434.
- Araujo, S.E.; Bernardo, W.M.; Habr-Gama, A.; Kiss, D.R.; Cecconello, I. DNA ploidy status and prognosis in colorectal cancer: a meta-analysis of published data. Colon Rectum. 2007, 50, 1800-1810. doi: 10.1007/s10350-007-9013-6.
- Crissman, J.D.; Zarbo, R.J.; Ma, C.K.; Visscher, D.W. Histopathologic parameters and DNA analysis in colorectal adenocarcinomas. Annu. 1989, 24, 103-147. PMID: 2671879.
- Michel, P.; Paresy, M.; Lepessot, F.; Hellot, M.F.; Paillot, B.; Scotte, M.; Peillon, C.; Ducrotté, P.; Hemet, J. Pre-operative kinetic parameter determination of colorectal adenocarcinomas. Prognostic significance. J. Gastroenterol. Hepatol. 2000, 12, 275-280. doi: 10.1097/00042737-200012030-00003.
- Zarbo, R.J.; Nakhleh, R.E.; Brown, R.D.; Kubus, J.J.; Ma, C.K.; Mackowiak, P. Prognostic significance of DNA ploidy and proliferation in 309 colorectal carcinomas as determined by two-color multiparametric DNA flow cytometry. Cancer. 1997, 79, 2073-2086. PMID: 9179053.
- Rew, D.A.; Wilson, G.D.; Taylor, I.; Weaver, P.C. Proliferation characteristics of human colorectal carcinomas measured in vivo. J. Surg. 1991, 78, 60-66. doi: 10.1002/bjs.1800780120.
- Gasinska, A.; Skolyszewski, J.; Popiela, T.; Richter, P.; Darasz, Z.; Nowak, K.; Niemiec, J.; Biesaga, B.; Adamczyk, A.; Bucki, K.; et al. Bromodeoxyuridine labeling index as an indicator of early tumor response to preoperative radiotherapy in patients with rectal cancer. Gastrointest. Surg. 2007, 11, 520-528. doi: 10.1007/s11605-007-0127-x.
- Magaki, S.; Hojat, S.A.; Wei, B.; So, A.; Yong, W.H. An Introduction to the Performance of Immunohistochemistry. Methods Mol. Biol. 2019, 1897, 289-298. doi: 10.1007/978-1-4939-8935-5_25.
- Mauriello, S.; Treglia, M.; Pallocci, M.; Bonfiglio, R.; Giacobbi, E.; Passalacqua, P.; Cammarano, A.; D'Ovidio, C.; Marsella, L.T.; Scimeca, M. Antigenicity Preservation Is Related to Tissue Characteristics and the Post-Mortem Interval: Immunohistochemical Study and Literature Review. Healthcare (Basel). 2022, 10, 1495. doi: 10.3390/healthcare10081495.
- Koo, M.; Squires, J.M.; Ying, D.; Huang, J. Making a Tissue Microarray. Methods Mol. Biol. 2019, 1897, 313-323. doi: 10.1007/978-1-4939-8935-5_27.
- Ye, C.; Wang, J.; Wu, P.; Li, X.; Chai, Y. Prognostic role of cyclin B1 in solid tumors: a meta-analysis. Oncotarget. 2017, 8, 2224-2232. doi: 10.18632/oncotarget.13653.
- Li, Y.; Wei, J.; Xu, C.; Zhao, Z.; You, T. Prognostic significance of cyclin D1 expression in colorectal cancer: a meta-analysis of observational studies. PLoS One. 2014, 9, e94508. doi: 10.1371/journal.pone.0094508.
- Yan, H.; Jiang, F.; Yang, J. Association of β-Catenin, APC, SMAD3/4, Tp53, and Cyclin D1 Genes in Colorectal Cancer: A Systematic Review and Meta-Analysis. Res. (Camb). 2022, 2022, 5338956. doi: 10.1155/2022/5338956.
- Zhou, H.; Huang, T.; Xiong, Y.; Peng, L.; Wang, R.; Zhang, G.J. The prognostic value of proliferating cell nuclear antigen expression in colorectal cancer: A meta-analysis. Medicine (Baltimore). 2018, 97, e13752. doi: 10.1097/MD.0000000000013752.
- Luo, Z.W.; Zhu, M.G.; Zhang, Z.Q.; Ye, F.J.; Huang, W.H.; Luo, X.Z. Increased expression of Ki-67 is a poor prognostic marker for colorectal cancer patients: a meta analysis. BMC Cancer. 2019, 19, 123. doi: 10.1186/s12885-019-5324-y.
- Duchrow, M.; Ziemann, T.; Windhövel, U.; Bruch, H.P.; Broll, R. Colorectal carcinomas with high MIB-1 labelling indices but low pKi67 mRNA levels correlate with better prognostic outcome. Histopathology. 2003, 42, 566-574. doi: 10.1046/j.1365-2559.2003.01613.x.
- Duchrow, M.; Häsemeyer, S.; Broll, R.; Bruch, H.P.; Windhövel, U. Assessment of proliferative activity in colorectal carcinomas by quantitative reverse transcriptase-polymerase chain reaction (RT-PCR). Cancer Invest. 2001, 19, 588-596. doi: 10.1081/cnv-100104286.
- Yue, S.Q.; Yang, Y.L.; Dou, K.F.; Li, K.Z. Expression of PCNA and CD44mRNA in colorectal cancer with venous invasion and its relationship to liver metastasis. World J. Gastroenterol. 2003, 9, 2863-2865. doi: 10.3748/wjg.v9.i12.2863.
- Cai, F.; Li, J.; Pan, X.; Zhang, C.; Wei, D.; Gao, C. Increased Expression of PCNA-AS1 in Colorectal Cancer and its Clinical Association. Lab. 2017, 63, 1809-1814. doi: 10.7754/Clin.Lab.2017.170503.
- Zhu, L.; Liu, J.; Ma, S.; Zhang, S. Long Noncoding RNA MALAT-1 Can Predict Metastasis and a Poor Prognosis: a Meta-Analysis. Oncol. Res. 2015, 21, 1259-1264. doi: 10.1007/s12253-015-9960-5.
- Perakis, S.O.; Thomas, J.E.; Pichler, M. Non-coding RNAs Enabling Prognostic Stratification and Prediction of Therapeutic Response in Colorectal Cancer Patients. Exp. Med. Biol. 2016, 937, 183-204. doi: 10.1007/978-3-319-42059-2_10.
- Yamada, A.; Yu, P.; Lin, W.; Okugawa, Y.; Boland, C.R.; Goel, A. A RNA-Sequencing approach for the identification of novel long non-coding RNA biomarkers in colorectal cancer. Rep. 2018, 8, 575. doi: 10.1038/s41598-017-18407-6.
- Chen, B.; Zhang, R.N.; Fan, X.; Wang, J.; Xu, C.; An, B.; Wang, Q.; Wang, J.; Leung, E.L.; Sui, X.; et al. Clinical diagnostic value of long non-coding RNAs in Colorectal Cancer: A systematic review and meta-analysis. Cancer. 2020, 11, 5518-5526. doi: 10.7150/jca.46358.
- Chen, S.; Shen, X. Long noncoding RNAs: functions and mechanisms in colon cancer. Cancer. 2020, 19, 167. doi: 10.1186/s12943-020-01287-2.
- Zhang, L.; Li, C.; Su, X. Emerging impact of the long noncoding RNA MIR22HG on proliferation and apoptosis in multiple human cancers. Exp. Clin. Cancer Res. 2020, 39, 271. doi: 10.1186/s13046-020-01784-8.
- Zhang, J.; Li, K.; Zheng, H.; Zhu, Y. Research progress review on long non-coding RNA in colorectal cancer. Neoplasma. 2021, 68, 240-252. doi: 10.4149/neo_2020_201012N1073.
- Lulli, M.; Napoli, C.; Landini, I.; Mini, E.; Lapucci, A. Role of Non-Coding RNAs in Colorectal Cancer: Focus on Long Non-Coding RNAs. J. Mol. Sci. 2022, 23, 13431. doi: 10.3390/ijms232113431.
- St Laurent, G.; Wahlestedt, C.; Kapranov, P. The Landscape of long noncoding RNA classification. Trends Genet. 2015, 31, 239-251. doi: 10.1016/j.tig.2015.03.007.
- Hombach, S.; Kretz, M. Non-coding RNAs: Classification, Biology and Functioning. Exp. Med. Biol. 2016, 937, 3-17. doi: 10.1007/978-3-319-42059-2_1.
- Takahashi, Y, Sawada, G, Kurashige, J, Uchi, R, Matsumura, T, Ueo, H, Takano, Y, Eguchi, H, Sudo, T, Sugimachi, K, et al. Amplification of PVT-1 is involved in poor prognosis via apoptosis inhibition in colorectal cancers. J. Cancer. 2014, 110, 164-171. doi: 10.1038/bjc.2013.698.
- To, K.K.; Tong, C.W.; Wu, M.; Cho, W.C. MicroRNAs in the prognosis and therapy of colorectal cancer: From bench to bedside. World J. Gastroenterol. 2018, 24, 2949-2973. doi: 10.3748/wjg.v24.i27.2949.
- Zhan, W.; Liao, X.; Chen, Z.; Li, L.; Tian, T.; Yu, L.; Li, R. LINC00858 promotes colorectal cancer by sponging miR-4766-5p to regulate PAK2. Cell Biol Toxicol. 2020, 36, 333-347. doi: 10.1007/s10565-019-09506-3.
- Zhan, Y.X.; Luo, G.H. DNA methylation detection methods used in colorectal cancer. World J. Clin. Cases. 2019, 7, 2916-2929. doi: 10.12998/wjcc.v7.i19.2916.
- Kothalawala, W.J.; Barták, B.K.; Nagy, Z.B.; Zsigrai, S.; Szigeti, K.A.; Valcz, G.; Takács, I.; Kalmár, A.; Molnár, B. A Detailed Overview About the Single-Cell Analyses of Solid Tumors Focusing on Colorectal Cancer. Oncol. Res. 2022, 28, 1610342. doi: 10.3389/pore.2022.1610342.
- Jones, T.; Townsend, D. History and future technical innovation in positron emission tomography. Med. Imaging (Bellingham). 2017, 4, 011013. doi: 10.1117/1.JMI.4.1.011013..
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Rev. Cell Dev. Biol. 2011, 27, 441-464. doi: 10.1146/annurev-cellbio-092910-154237.
- Pelosi, E, Deandreis, D. The role of 18F-fluoro-deoxy-glucose positron emission tomography (FDG-PET) in the management of patients with colorectal cancer. J. Surg. Oncol. 2007, 33, 1-6. doi: 10.1016/j.ejso.2006.10.020.
- Flamen, P.; Stroobants, S.; Van Cutsem, E.; Dupont, P.; Bormans, G.; De Vadder, N.; Penninckx, F.; Van Hoe, L.; Mortelmans, L. Additional value of whole-body positron emission tomography with fluorine-18-2-fluoro-2-deoxy-D-glucose in recurrent colorectal cancer. Clin. Oncol. 1999, 17, 894-901. doi: 10.1200/JCO.1999.17.3.894.
- Yamamoto, Y.; Kameyama, R.; Izuishi, K.; Takebayashi, R.; Hagiike, M.; Asakura, M.; Haba, R.; Nishiyama, Y. Detection of colorectal cancer using ¹⁸F-FLT PET: comparison with ¹⁸F-FDG PET. Med. Commun. 2009, 30, 841-845. doi: 10.1097/MNM.0b013e328330294d.
- Deng, S.M.; Zhang, W.; Zhang, B.; Chen, Y.Y.; Li, J.H.; Wu, Y.W. Correlation between the Uptake of 18F-Fluorodeoxyglucose (18F-FDG) and the Expression of Proliferation-Associated Antigen Ki-67 in Cancer Patients: A Meta-Analysis. PLoS One. 2015, 10, e0129028. doi: 10.1371/journal.pone.0129028.
This entry is adapted from the peer-reviewed paper 10.3390/cancers15184570