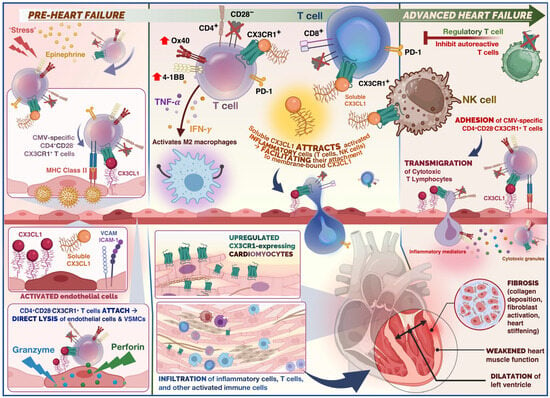
Dilated cardiomyopathy (DCM) is a cardiac condition with structural and functional impairment, where either the left ventricle or both ventricular chambers are enlarged, coinciding with reduced systolic pump function (reduced ejection fraction, rEF). The prevalence of DCM is more than 1:250 individuals, and mortality largely due to heart failure in two-third of cases, and sudden cardiac death in one-third of patients. Damage to the myocardium, whether from a genetic or environmental cause such as viruses, triggers inflammation and recruits immune cells to the heart to repair the myocardium. Examination of myocardial biopsy tissue often reveals an inflammatory cell infiltrate, T lymphocyte (T cell) infiltration, or other activated immune cells. Despite medical therapy, adverse outcomes for DCM remain. The evidence base and existing literature suggest that upregulation of CX3CR1, migration of immune cells, together with cytomegalovirus (CMV) seropositivity is associated with worse outcomes in patients with dilated cardiomyopathy.
1. Introduction
A reduced ejection fraction, or HFrEF, and enlargement of either the left ventricle or both ventricular chambers are hallmarks of dilated cardiomyopathy (DCM), a disorder that affects both the structure and function of the heart. Clinically, patients display heart failure symptoms in the absence of congenital, valvular, or coronary artery disease. DCM is categorised as either inherited (familial) or acquired (non familial) by the European Society of Cardiology (ESC). Male patients experience DCM more frequently (2:1 to 3:1) and with worse outcomes than female patients [
1]. More than 1 in 250 people have DCM; progressive heart failure (70%) and sudden cardiac death (30%) account for the majority of deaths [
2]. There is still a significant mortality rate even with recent improvements in the guideline-recommended heart failure treatment. Remodelling and fibrosis leading to the development of scar tissue promote the distortion of the ventricle into a spherical shape, causing the chambers to dilate, and as a result, the stroke volume, cardiac output, ventricular filling, and left ventricular end diastolic pressure (LVEDP) are all affected negatively. Early intervention is difficult before permanent damage has taken place since heart failure symptoms frequently appear gradually after the beginning of cardiac damage.
Damage to the myocardium, whether caused by genetic or environmental factors, induces inflammation and recruits immune cells to repair the myocardium; the most common causes of inflammatory DCM are infections and autoimmunity [
3]. Examining myocardial biopsy tissue typically reveals an inflammatory cell infiltrate, such as CD3 T lymphocyte infiltration, or other activated immune cells, such as M2 macrophages and B lymphocytes, in autoimmune diseases such as rheumatoid arthritis, systemic sclerosis, systemic lupus erythematosus, and Dresslers syndrome to name a few. All of these are capable of releasing cytokines, including transforming growth factor-1 (TGF1), interleukin-4 (IL-4), IL-1, IL-17A, IL-33, and tumour necrosis factor (TNF), which can promote remodelling, collagen deposition, and fibrosis [
4]. Fibrosis results from inflammation at the site of tissue injury and is the distinguishing pathological feature of DCM, in addition to dilatation [
5] (see
Figure 1).
Figure 1. Summary of the role of immune cells in the cardiovascular disease continuum from the pre-heart failure to advanced heart failure stage in dilated cardiomyopathy (DCM). Physiological stress and release of epinephrine in the acute phase (pre-heart failure), triggers release of CX3CR1+ T cells. In the case of stress-induced viral reactivation, rapid mobilisation for CMV-specific T cells occurs. CX3CR1+ CMV-specific CD4+ T cells would adhere to endothelium, and undergo antigen recognition, leading to activation of cytotoxic effector cells. Direct lysis of endothelial cells and vascular smooth muscle cells (VSMCs) occurs (mediated by granzymes and perforin). Release of cytokines such as TNF-α and IFN-γ as well as chemokines from CMV-specific CD4+ T cells potentially attracts further immune cells such as polarised macrophages and NK cells through endothelial induction of fractalkine, which may enhance endothelial injury. The CX3CR1– regulatory T cells (Tregs) are capable of inhibiting autoreactive T cells. Thus, administration of Tregs serves as a potential therapeutic target to promote immune tolerance. Soluble fractalkine binds to its chemokine receptor on cytotoxic T cells and NK cells which facilitates adhesion and transmigration across the endothelium. Infiltrating T cells and inflammatory cells release inflammatory mediators and cytotoxic granules which promote remodelling, collagen deposition, and fibrosis. This ultimately results in weakening of the ventricular heart muscle and ventricular dilatation.
2. Phenotypic Clustering of Dilated Cardiomyopathy Patients
Recent research from the Maastricht Cardiomyopathy Registry included 795 consecutive DCM patients who underwent extensive phenotyping, including extensive clinical data on aetiology and comorbidities, imaging, and endomyocardial biopsies [
9]. On the basis of unsupervised hierarchical clustering of principal components, the authors found four mutually exclusive and clinically distinct phenogroups (PG):
-
PG1 included 331 patients with mild systolic dysfunction
-
(Mean ejection fraction: 43% ± 9%),
-
PG2 included 83 patients with auto-immune disease background
-
PG3 included 165 patients with cardiac arrhythmias (mainly atrial fibrillation and ventricular tachycardias), including patients with genetic causes (Familial cardiomyopathy)
-
PG4 included 216 patients with severe systolic dysfunction (Mean ejection fraction: 23% ± 8%)
RNA-sequencing of cardiac biopsy samples (n = 91) revealed a unique molecular profile per PG: pro-inflammatory (PG2, auto-immune), pro-fibrotic (PG3, arrhythmia), and metabolic (PG4, low EF) gene expression. In addition, event-free survival varied between the four phenogroups, even after adjustment for well-known clinical predictors. Using decision tree modelling, four clinical parameters (autoimmune disease, ejection fraction, atrial fibrillation, and renal function) were identified by which every DCM patient from two independent DCM cohorts could be assigned to one of the four phenogroups with corresponding outcomes.
3. Aetiologies of Dilated Cardiomyopathy
3.1. Familial Cardiomyopathy
Although a genetic basis for familial DCM is well established, the majority of cases of DCM appear to be sporadic; that is, even when the family members of patients newly diagnosed with idiopathic DCM are clinically screened, the majority of family members have no evidence of DCM; thus, patients are ultimately diagnosed with nonfamilial (sporadic) DCM. No large, multicentre study of families whose members have been systematically clinically screened for DCM and also undergone exome or genome sequencing to identify a potential genetic cause has been published to date. A total of 15–30% of patients with DCM may be diagnosed with familial DCM if their family members undertake clinical screening, according to family-based studies [
3]. Historically, large multigenerational familial DCM pedigrees have served as the foundation for the majority of DCM-associated gene discovery. These multigenerational genealogies have provided statistically sound genetic evidence for the causality of variants in DCM-associated genes.
3.2. Autoimmune Myocarditis
Autoimmune myocarditis is a condition in which cardiac muscle is damaged by self-reactive immune cells. This condition is typically associated with numerous genetic and environmental risk factors. Various systemic autoimmune diseases, such as sarcoidosis or SLE (lupus), can compromise cardiac function by a variety of mechanisms [
11]. The prognosis for patients with either of these conditions is typically dismal. However, it has been demonstrated that early administration of glucocorticosteroids and immunosuppressive agents improves patients’ conditions [
12]. Restoring the equilibrium between autoimmunity and immune tolerance, which is regulated by Th17 and regulatory T cells (Tregs), is one of the primary therapeutic approaches for autoimmune disease [
13]. Treg cells are capable of inhibiting autoreactive T cells, and multiple studies have repeatedly identified a deficiency in the quantity or function of Treg cells in autoimmune disorders [
13]. Therefore, Treg administration has been demonstrated to be a promising treatment for a variety of autoimmune disorders.
3.3. Post-Viral Myocarditis
Myocarditis is frequently preceded by infection, with viruses being the most prevalent cause of this condition [
15,
16]. There are typically three phases of viral myocarditis. During the first phase, which lasts several days, the virus obtains entry and actively replicates. Therefore, the innate immune response will be triggered against these exogenous substances. During this phase, direct viral burden contributes significantly to myocardial damage. The subsequent phase revealed that an excessive immune response is the leading cause of cardiac damage. During this phase, T cell infiltration has been reported alongside an increase in fibrosis and calcification of the myocardium. In the final phase, patients may experience either remission or progression to dilated cardiomyopathy (DCM), depending on the heart’s capacity to recover from previous insults of direct viral injury and immune persistence [
15,
16].
3.4. Immune Checkpoint Inhibitor-Related Myocarditis
Immune checkpoint inhibitors (ICIs) are a novel type of cancer treatment that is being applied to a growing number of cancer types for example malignant melanoma, Hodgkin’s lymphoma and non small cell lung cancer to name a few. The immune modulators CTLA-4, PD-1, and PD-L1 are the targets of ICIs [
19]. ICIs may, however, stimulate T cell activity against host tissues, leading to immune-related adverse events (irAEs). Myocarditis is a rare adverse reaction associated with ICIs, with incidence rates ranging from 0.1% to 2% [
20]. With ICI myocarditis, there is a reduction in absolute lymphocyte count and an increase in neutrophils, both of which are associated with subsequent significant adverse cardiac events [
20]. Cardiovascular magnetic resonance (CMR) utilising tissue characterisation techniques such as late gadolinium enhancement (LGE) and the presence of myocardial oedema, is the gold standard non-invasive imaging test for diagnosis and risk prediction in myocarditis of other aetiologies.
CTLA-4 is a crucial regulator of T cell inhibition via multiple mechanisms, including negative signalling of B7-CD28, inhibition of IL-2 mRNA expression, and interaction with the TCR-CD3 pathway to inhibit T cell activation [
22,
23]. After IFN-g exposure, PD-1 and PD-L1 were highly expressed to prevent T-cell-mediated signalling on host cells. The FDA has currently approved seven ICI medications for cancer therapy. Although ICI-associated myocarditis is uncommon, its mortality rate can reach 25 to 50 percent [
23]. In addition, this adverse event was reported to occur within 1 to 2 months of the initial ICI dose. The underlying pathogenesis of ICI-associated myocarditis remains unknown.
4. Role of Fractalkine Signalling in Cardiovascular Disease
4.1. Fractalkine (CX3CL1) and Its Receptor CX3CR1 in Atherogenesis
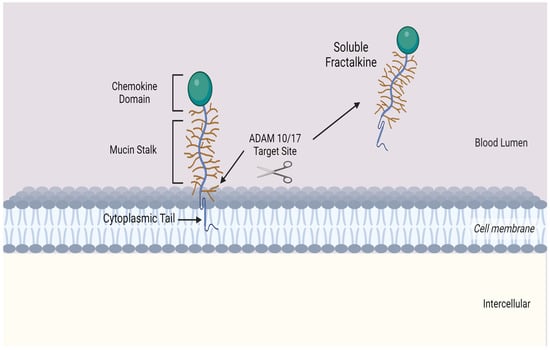
Fractalkine consists of 373 amino acids and is the only member of the CX
3C chemokine subfamily with both membrane-bound and soluble variants [
25,
26,
27,
28]. The former is an adhesion molecule with four sections: an extracellular N-terminal domain, a mucin-like stalk, a transmembrane alpha helix, and a brief cytoplasmic tail. Fractalkine can be cleaved at the juncture of its stalk and transmembrane helix my metalloproteinases ADAM 10 or ADAM 17 to produce its soluble form, which functions as a chemoattractant (see
Figure 2) for monocytes, NK cells, and T cells.
Figure 2. Transmembrane chemokine Fractalkine (CX3CL1). Graphic illustration of the transmembrane chemokine. It contains an extracellular N-domain, mucin stalk and a short cytoplasmic trail. The soluble form is generated by shedding and cleavage by metalloproteinases ADAM 10 and ADAM 17. The resulting release of the mucin-like stalk and chemokine domain acts as a chemoattractant for inflammatory cells.].
4.2. CX3CR1 and Myocardial Infarction
The CX
3CR1 receptor is essential for the recruitment of lymphocytes that contribute to cardiac ischaemia/reperfusion injury and subsequent adverse remodelling. A subset of lymphocytes with cytotoxic properties adhere and are marginalised upon interaction with the fractalkine receptor CX
3CR1 expressed on circulating lymphocytes (see
Figure 3).
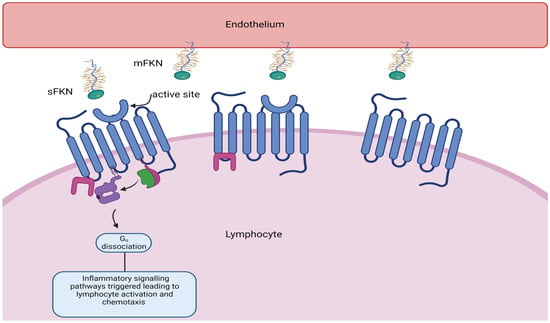
Figure 3. Soluble fractalkine (CX3CL1) binds to CX3CR1, its G-coupled receptor, resulting in lymphocyte activation adhesion and migration across the endothelium, resulting in tissue damage.
4.3. Fractalkine Signalling in Heart Failure
In a study conducted by Nakayama and colleagues, higher myocardial immune activation was found to be associated with a poor prognosis for DCM [
8]. Patients with higher counts of CD3
−, CD68
−, and CD163-positive infiltrating cells had substantially worse outcomes (
p = 0.007,
p = 0.011, and
p = 0.022, respectively). The association between M2 macrophages and collagen synthesis suggests that ventricular remodelling in DCM may be associated with the phenotypic polarisation of macrophages towards M2. CX
3CR1 and CX
3CL1 expression is consistently upregulated in cardiac tissue from heart failure patients with various aetiologies, including end-stage DCM. CX
3CR1 and CX
3CL1 gene expression has been found to be elevated in both DCM and ICM patient cardiac tissue [
32]. Richter and coworkers measured the plasma levels of CX
3CL1 in 349 patients with advanced systolic heart failure [
53].
5. Link between CX3CR1 and Cytomegalovirus
Cytomegalovirus (CMV) is a pervasive herpes virus with a seroprevalence of greater than 60% in individuals aged 50 and older in the majority of studies [
54] and 85% in those aged 80 and older [
46]. CMV causes an asymptomatic or moderate initial response in immunocompetent hosts, but it is never eliminated from the body, resulting in lifelong latent infection with the possibility of reactivation [
54]. Seropositivity for CMV induces significant changes in the T cell composition of the host. CMV-specific CD8
+ and, to a lesser extent, CD4
+ memory T cells, particularly TEMRA cells, account for a disproportionately high proportion of total T cells; this phenomenon is known as memory inflation [
55,
56,
57]. There is evidence that memory expansion in the CD8 compartment is greater in older patients, indicating that the CMV-specific memory population continues to expand with age, but this is less evident and has been less thoroughly studied in CD4 T cells [
48]. Memory inflation may be an adaptive response to suppress reactivations [
58], and whether this unbalanced expansion impedes the immune system’s function remains debatable, as some contend that the CD8 compartment is sufficiently plastic to permit expansion of one phenotype without disruption of others [
59]. In the presence of viral antigen, CMV-specific CD4
+ T cells have been shown to cause endothelial injury, with more damage occurring in donors with higher frequencies of CMV-specific CD4
+ T cells [
60,
61,
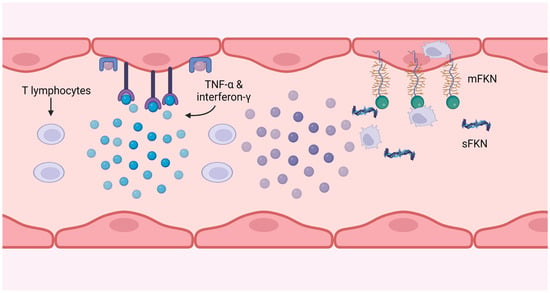
62]. This injury is caused by the release of IFN-g and TNF-α by T cells at sufficient levels to induce endothelial cell induction of fractalkine, thereby attracting natural killer (NK) cells and monocyte-macrophages, as depicted in
Figure 4.
Figure 4. CMV-specific T cells produce interferon-g and TNF-α, which induce fractalkine expression on the endothelium. The soluble form attracts monocytes and NK cells, and the membrane-bound form catches and internalises them, removing them from the circulation.
6. Conclusions
The evidence base and existing literature suggest that upregulation of fractalkine/CX3CR1, together with CMV seropositivity and consequent migration of immune and inflammatory cells, is associated with worse outcomes in patients with inflammatory conditions including dilated cardiomyopathy. We hypothesise that dilated cardiomyopathy, whether acquired secondary to infection, autoimmunity, drugs and toxins or familial in origin, may lead to cardiac inflammation and fibrosis, resulting in adverse remodelling through the interplay of these factors. Robust observational studies are required to identify a subgroup of at-risk patients with dilated cardiomyopathy through in-depth phenomapping, genotyping, evaluation of immune status, and viral presence, which in turn may inform future drug trials utilising immune modulators to target this pathway.
This entry is adapted from the peer-reviewed paper 10.3390/cells12192377