Avascular necrosis of the femoral head (ANFH) is a painful disorder characterized by the cessation of blood supply to the femoral head, leading to its death and subsequent joint collapse. Influenced by several risk factors, including corticosteroid use, excessive alcohol intake, hypercholesterolemia, smoking and some inflammatory disorders, along with cancer, its clinical consequences are thrombus formation due to underlying inflammation and endothelial dysfunction, which collaborates with coagulopathy and impaired angiogenesis. Nonetheless, angiogenesis resolves the obstructed free flow of the blood by providing alternative routes. Clinical manifestations of early stage of ANFH mimic cysts or lesions in subchondral bone, vasculitis and transient osteoporosis of the hip, rendering it difficult to diagnose, complex to understand and complicated to cure.
- avascular necrosis of femoral head
- impaired angiogenesis
- coagulopathy
- endothelial dysfunction
- bone disease
- skeletal abnormality
1. Introduction
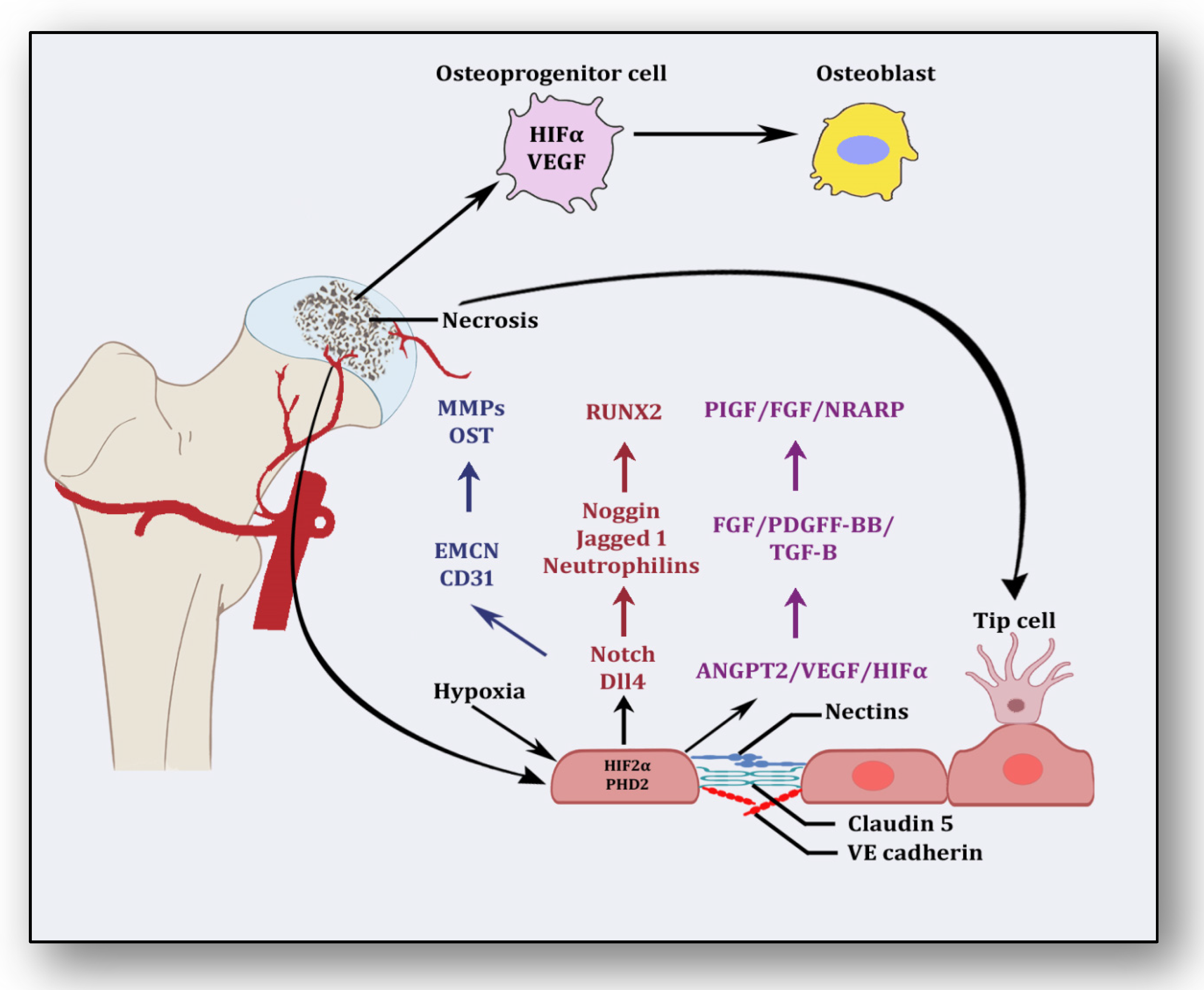
2. Angiogenesis: Sprouting, Splitting and Stabilization
3. Angiogenesis: A Predominant Pacifier in Avascular Necrosis
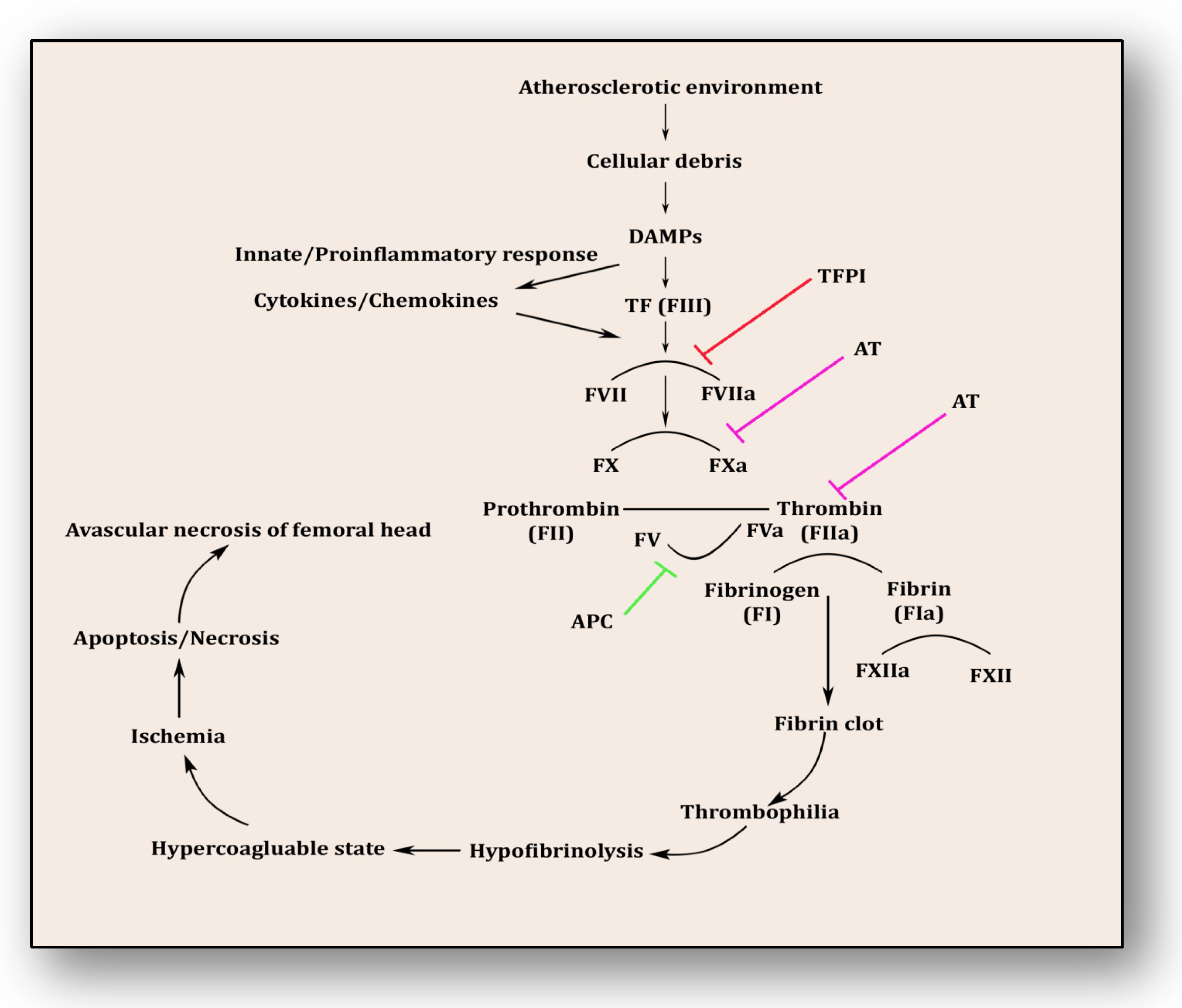
4. Coagulopathy: A Culprit Alliance of Thrombophilia and Hypofibrinolysis
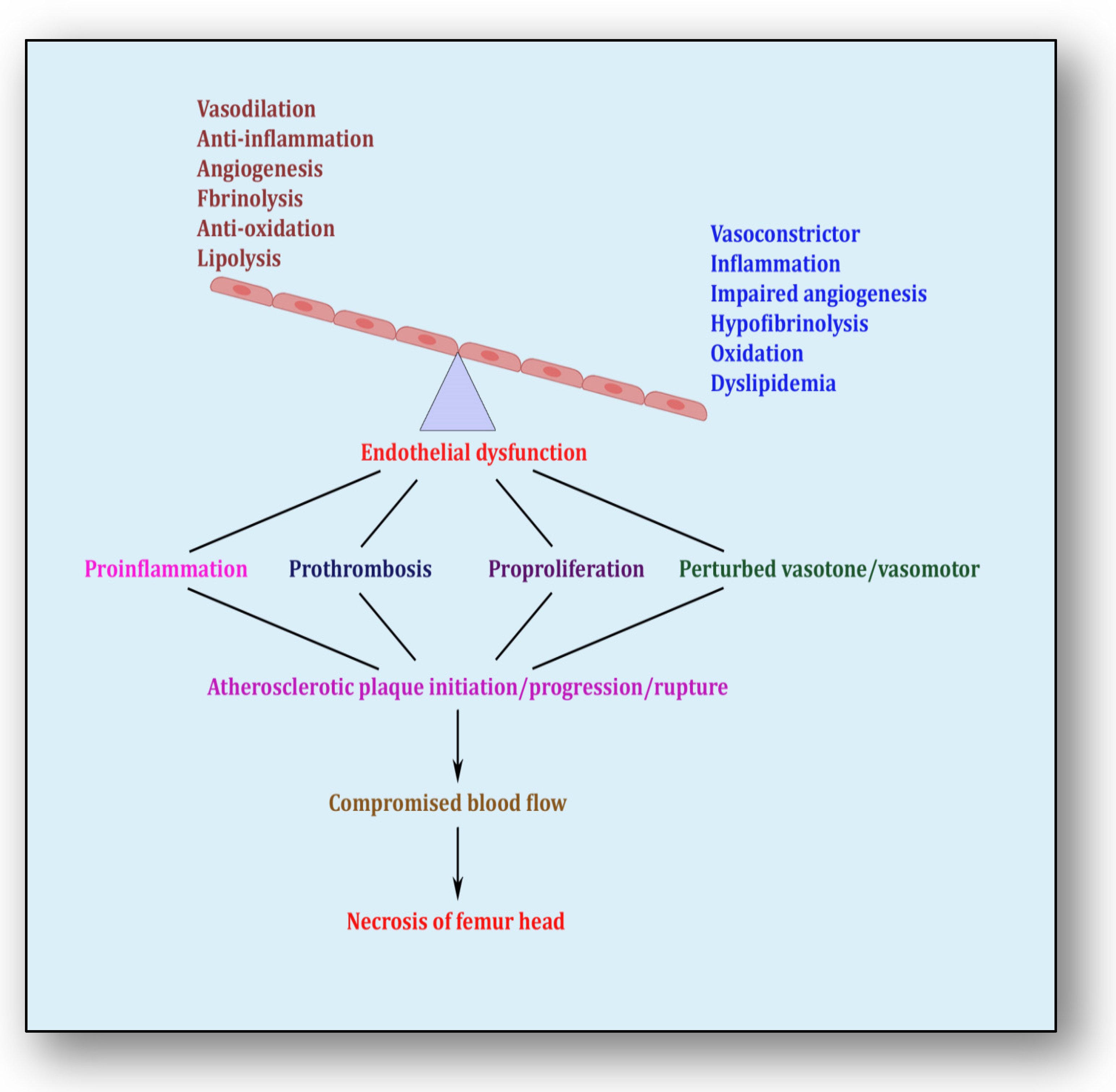
5. Endothelial Dysfunction: Holding Hands with Inflammation
6. Therapies Used in Other Diseases: A Possible Avenue for ANFH Management
This entry is adapted from the peer-reviewed paper 10.3390/cells12182278
References
- Narayanan, A.; Khanchandani, P.; Borkar, R.M.; Ambati, C.R.; Roy, A.; Han, X.; Bhoskar, R.N.; Ragampeta, S.; Gannon, F.; Mysorekar, V.; et al. Avascular Necrosis of Femoral Head: A Metabolomic, Biophysical, Biochemical, Electron Microscopic and Histopathological Characterization. Sci. Rep. 2017, 7, 10721.
- George, G.; Lane, J.M. Osteonecrosis of the Femoral Head. J. Am. Acad. Orthop. Surg. Glob. Res. Rev. 2022, 6, e21.00176.
- Petek, D.; Hannouche, D.; Suva, D. Osteonecrosis of the Femoral Head: Pathophysiology and Current Concepts of Treatment. EFORT Open Rev. 2019, 4, 85–97.
- Kerachian, M.A.; Harvey, E.J.; Cournoyer, D.; Chow, T.Y.K.; Séguin, C. Avascular Necrosis of the Femoral Head: Vascular Hypotheses. Endothelium 2006, 13, 237–244.
- Kretschmer, M.; Rüdiger, D.; Zahler, S. Mechanical Aspects of Angiogenesis. Cancers 2021, 13, 4987.
- Semenza, G.L. Vasculogenesis, Angiogenesis, and Arteriogenesis: Mechanisms of Blood Vessel Formation and Remodeling. J. Cell. Biochem. 2007, 102, 840–847.
- Ashraf, J.V.; Al Haj Zen, A. Role of Vascular Smooth Muscle Cell Phenotype Switching in Arteriogenesis. Int. J. Mol. Sci. 2021, 22, 10585.
- Claesson-Welsh, L. Vascular Permeability--the Essentials. Upsala J. Med. Sci. 2015, 120, 135–143.
- Stratman, A.N.; Davis, G.E. Endothelial Cell-Pericyte Interactions Stimulate Basement Membrane Matrix Assembly: Influence on Vascular Tube Remodeling, Maturation, and Stabilization. Microsc. Microanal. 2012, 18, 68–80.
- Yoo, S.Y.; Kwon, S.M. Angiogenesis and Its Therapeutic Opportunities. Mediat. Inflamm. 2013, 2013, 127170.
- Smith, D.W. Is Avascular Necrosis of the Femoral Head the Result of Inhibition of Angiogenesis? Med. Hypotheses 1997, 49, 497–500.
- Wu, S.-H.; Miao, Y.; Zhu, X.-Z.; Li, G.-Y. . Zhongguo Gu Shang 2023, 36, 294–298.
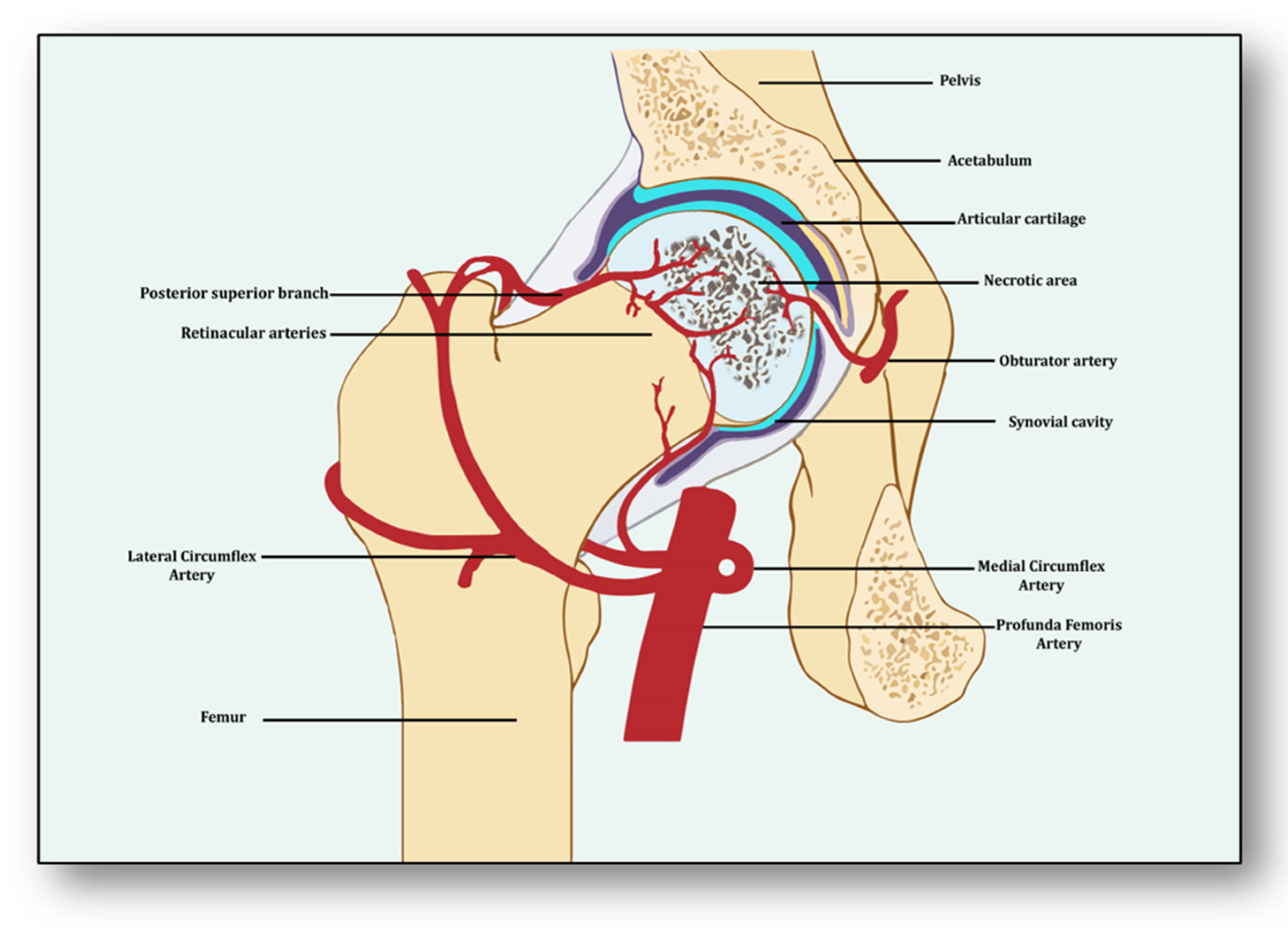
- Kawasaki, Y.; Kinose, S.; Kato, K.; Sakai, T.; Ichimura, K. Anatomic Characterization of the Femoral Nutrient Artery: Application to Fracture and Surgery of the Femur. Clin. Anat. 2020, 33, 479–487.
- Rajani, S.J.; Ravat, M.K.; Rajani, J.K.; Bhedi, A.N. Cadaveric Study of Profunda Femoris Artery with Some Unique Variations. J. Clin. Diagn. Res. 2015, 9, AC01–AC03.
- Zhao, D.; Qiu, X.; Wang, B.; Wang, Z.; Wang, W.; Ouyang, J.; Silva, R.M.; Shi, X.; Kang, K.; Xu, D.; et al. Epiphyseal Arterial Network and Inferior Retinacular Artery Seem Critical to Femoral Head Perfusion in Adults With Femoral Neck Fractures. Clin. Orthop. Relat. Res. 2017, 475, 2011–2023.
- Lee, E.-J.; Jain, M.; Alimperti, S. Bone Microvasculature: Stimulus for Tissue Function and Regeneration. Tissue Eng. Part B Rev. 2021, 27, 313–329.
- Grüneboom, A.; Kling, L.; Christiansen, S.; Mill, L.; Maier, A.; Engelke, K.; Quick, H.H.; Schett, G.; Gunzer, M. Next-Generation Imaging of the Skeletal System and Its Blood Supply. Nat. Rev. Rheumatol. 2019, 15, 533–549.
- Grüneboom, A.; Hawwari, I.; Weidner, D.; Culemann, S.; Müller, S.; Henneberg, S.; Brenzel, A.; Merz, S.; Bornemann, L.; Zec, K.; et al. A Network of Trans-Cortical Capillaries as Mainstay for Blood Circulation in Long Bones. Nat. Metab. 2019, 1, 236–250.
- Kusumbe, A.P.; Ramasamy, S.K.; Adams, R.H. Coupling of Angiogenesis and Osteogenesis by a Specific Vessel Subtype in Bone. Nature 2014, 507, 323–328.
- Xu, Z.; Kusumbe, A.P.; Cai, H.; Wan, Q.; Chen, J. Type H Blood Vessels in Coupling Angiogenesis-Osteogenesis and Its Application in Bone Tissue Engineering. J. Biomed. Mater. Res. B Appl. Biomater. 2023, 111, 1434–1446.
- Liu, Y.; Xie, H.-Q.; Shen, B. Type H Vessels-a Bridge Connecting Subchondral Bone Remodelling and Articular Cartilage Degeneration in Osteoarthritis Development. Rheumatology 2023, 62, 1436–1444.
- Zheng, G.-S.; Qiu, X.; Wang, B.-J.; Zhao, D.-W. Relationship Between Blood Flow and Collapse of Nontraumatic Osteonecrosis of the Femoral Head. J. Bone Jt. Surg. Am. 2022, 104, 13–18.
- Muñoz-Chápuli, R.; Quesada, A.R.; Angel Medina, M. Angiogenesis and Signal Transduction in Endothelial Cells. Cell. Mol. Life Sci. 2004, 61, 2224–2243.
- Kazerounian, S.; Lawler, J. Integration of Pro- and Anti-Angiogenic Signals by Endothelial Cells. J. Cell Commun. Signal. 2018, 12, 171–179.
- Ding, H.; Gao, Y.-S.; Hu, C.; Wang, Y.; Wang, C.-G.; Yin, J.-M.; Sun, Y.; Zhang, C.-Q. HIF-1α Transgenic Bone Marrow Cells Can Promote Tissue Repair in Cases of Corticosteroid-Induced Osteonecrosis of the Femoral Head in Rabbits. PLoS ONE 2013, 8, e63628.
- Felmeden, D.C.; Blann, A.D.; Lip, G.Y.H. Angiogenesis: Basic Pathophysiology and Implications for Disease. Eur. Heart J. 2003, 24, 586–603.
- Han, Y.; You, X.; Xing, W.; Zhang, Z.; Zou, W. Paracrine and Endocrine Actions of Bone-the Functions of Secretory Proteins from Osteoblasts, Osteocytes, and Osteoclasts. Bone Res. 2018, 6, 16.
- Dor, Y.; Keshet, E. Ischemia-Driven Angiogenesis. Trends Cardiovasc. Med. 1997, 7, 289–294.
- Corrado, C.; Fontana, S. Hypoxia and HIF Signaling: One Axis with Divergent Effects. Int. J. Mol. Sci. 2020, 21, 5611.
- Hu, K.; Olsen, B.R. The Roles of Vascular Endothelial Growth Factor in Bone Repair and Regeneration. Bone 2016, 91, 30–38.
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011, 2, 1097–1105.
- Asahara, T.; Takahashi, T.; Masuda, H.; Kalka, C.; Chen, D.; Iwaguro, H.; Inai, Y.; Silver, M.; Isner, J.M. VEGF Contributes to Postnatal Neovascularization by Mobilizing Bone Marrow-Derived Endothelial Progenitor Cells. EMBO J. 1999, 18, 3964–3972.
- Yang, Y.-Q.; Tan, Y.-Y.; Wong, R.; Wenden, A.; Zhang, L.-K.; Rabie, A.B.M. The Role of Vascular Endothelial Growth Factor in Ossification. Int. J. Oral Sci. 2012, 4, 64–68.
- Zelzer, E.; Mamluk, R.; Ferrara, N.; Johnson, R.S.; Schipani, E.; Olsen, B.R. VEGFA Is Necessary for Chondrocyte Survival during Bone Development. Development 2004, 131, 2161–2171.
- Radke, S.; Battmann, A.; Jatzke, S.; Eulert, J.; Jakob, F.; Schütze, N. Expression of the Angiomatrix and Angiogenic Proteins CYR61, CTGF, and VEGF in Osteonecrosis of the Femoral Head. J. Orthop. Res. 2006, 24, 945–952.
- Zhang, C.; Li, Y.; Cornelia, R.; Swisher, S.; Kim, H. Regulation of VEGF Expression by HIF-1α in the Femoral Head Cartilage Following Ischemia Osteonecrosis. Sci. Rep. 2012, 2, 650.
- Street, J.; Bao, M.; deGuzman, L.; Bunting, S.; Peale, F.V.; Ferrara, N.; Steinmetz, H.; Hoeffel, J.; Cleland, J.L.; Daugherty, A.; et al. Vascular Endothelial Growth Factor Stimulates Bone Repair by Promoting Angiogenesis and Bone Turnover. Proc. Natl. Acad. Sci. USA 2002, 99, 9656–9661.
- Hu, K.; Olsen, B.R. Osteoblast-Derived VEGF Regulates Osteoblast Differentiation and Bone Formation during Bone Repair. J. Clin. Investig. 2016, 126, 509–526.
- Kapitsinou, P.P.; Rajendran, G.; Astleford, L.; Michael, M.; Schonfeld, M.P.; Fields, T.; Shay, S.; French, J.L.; West, J.; Haase, V.H. The Endothelial Prolyl-4-Hydroxylase Domain 2/Hypoxia-Inducible Factor 2 Axis Regulates Pulmonary Artery Pressure in Mice. Mol. Cell. Biol. 2016, 36, 1584–1594.
- Gavard, J.; Gutkind, J.S. VE-Cadherin and Claudin-5: It Takes Two to Tango. Nat. Cell Biol. 2008, 10, 883–885.
- Carmeliet, P.; Jain, R.K. Molecular Mechanisms and Clinical Applications of Angiogenesis. Nature 2011, 473, 298–307.
- Hutchings, H.; Ortega, N.; Plouët, J. Extracellular Matrix-Bound Vascular Endothelial Growth Factor Promotes Endothelial Cell Adhesion, Migration, and Survival through Integrin Ligation. FASEB J. 2003, 17, 1520–1522.
- Phng, L.-K.; Potente, M.; Leslie, J.D.; Babbage, J.; Nyqvist, D.; Lobov, I.; Ondr, J.K.; Rao, S.; Lang, R.A.; Thurston, G.; et al. Nrarp Coordinates Endothelial Notch and Wnt Signaling to Control Vessel Density in Angiogenesis. Dev. Cell 2009, 16, 70–82.
- Bergers, G.; Song, S. The Role of Pericytes in Blood-Vessel Formation and Maintenance. Neuro-Oncology 2005, 7, 452–464.
- Noel, A.; Maillard, C.; Rocks, N.; Jost, M.; Chabottaux, V.; Sounni, N.E.; Maquoi, E.; Cataldo, D.; Foidart, J.M. Membrane Associated Proteases and Their Inhibitors in Tumour Angiogenesis. J. Clin. Pathol. 2004, 57, 577–584.
- Vosmaer, A.; Pereira, R.R.; Koenderman, J.S.; Rosendaal, F.R.; Cannegieter, S.C. Coagulation Abnormalities in Legg-Calvé-Perthes Disease. J. Bone Jt. Surg. Am. 2010, 92, 121–128.
- Dahlbäck, B.; Carlsson, M.; Svensson, P.J. Familial Thrombophilia Due to a Previously Unrecognized Mechanism Characterized by Poor Anticoagulant Response to Activated Protein C: Prediction of a Cofactor to Activated Protein C. Proc. Natl. Acad. Sci. USA 1993, 90, 1004–1008.
- Svensson, P.J.; Dahlbäck, B. Resistance to Activated Protein C as a Basis for Venous Thrombosis. N. Engl. J. Med. 1994, 330, 517–522.
- Molino, D.; De Santo, N.G.; Marotta, R.; Anastasio, P.; Mosavat, M.; De Lucia, D. Plasma Levels of Plasminogen Activator Inhibitor Type 1, Factor VIII, Prothrombin Activation Fragment 1+2, Anticardiolipin, and Antiprothrombin Antibodies Are Risk Factors for Thrombosis in Hemodialysis Patients. Semin. Nephrol. 2004, 24, 495–501.
- Zwaginga, J.J.; Ijsseldijk, M.J.; Beeser-Visser, N.; de Groot, P.G.; Vos, J.; Sixma, J.J. High von Willebrand Factor Concentration Compensates a Relative Adhesion Defect in Uremic Blood. Blood 1990, 75, 1498–1508.
- Fortmann, S.P.; Marcovina, S.M. Lipoprotein(a), a Clinically Elusive Lipoprotein Particle. Circulation 1997, 95, 295–296.
- Hughes, G.R. The Antiphospholipid Syndrome: Ten Years On. Lancet 1993, 342, 341–344.
- Mont, M.A.; Jones, L.C.; Hungerford, D.S. Nontraumatic Osteonecrosis of the Femoral Head: Ten Years Later. J. Bone Jt. Surg. Am. 2006, 88, 1117–1132.
- Dubois, E.L.; Cozen, L. Avascular (Aseptic) Bone Necrosis Associated with Systemic Lupus Erythematosus. JAMA 1960, 174, 966–971.
- Ware, H.E.; Brooks, A.P.; Toye, R.; Berney, S.I. Sickle Cell Disease and Silent Avascular Necrosis of the Hip. J. Bone Jt. Surg. Br. 1991, 73, 947–949.
- Sondag, D.; Verhoeven, S.; Löwik, D.W.P.M.; van Geffen, M.; Veer, C.V.; van Heerde, W.L.; Boltje, T.J.; Rutjes, F.P.J.T. Activity Sensing of Coagulation and Fibrinolytic Proteases. Chemistry 2023, 29, e202203473.
- Noubouossie, D.F.; Reeves, B.N.; Strahl, B.D.; Key, N.S. Neutrophils: Back in the Thrombosis Spotlight. Blood 2019, 133, 2186–2197.
- Butenas, S.; Orfeo, T.; Mann, K.G. Tissue Factor in Coagulation: Which? Where? When? Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1989–1996.
- Palta, S.; Saroa, R.; Palta, A. Overview of the Coagulation System. Indian J. Anaesth. 2014, 58, 515–523.
- Schmitz, T.; Bäuml, C.A.; Imhof, D. Inhibitors of Blood Coagulation Factor XIII. Anal. Biochem. 2020, 605, 113708.
- Periayah, M.H.; Halim, A.S.; Mat Saad, A.Z. Mechanism Action of Platelets and Crucial Blood Coagulation Pathways in Hemostasis. Int. J. Hematol. Oncol. Stem Cell Res. 2017, 11, 319–327.
- Sachs, U.J.; Kirsch-Altena, A.; Müller, J. Markers of Hereditary Thrombophilia with Unclear Significance. Hamostaseologie 2022, 42, 370–380.
- Meager, A. Cytokine Regulation of Cellular Adhesion Molecule Expression in Inflammation. Cytokine Growth Factor Rev. 1999, 10, 27–39.
- Ito, T. PAMPs and DAMPs as Triggers for DIC. J. Intensive Care 2014, 2, 67.
- Glueck, C.J.; Freiberg, R.A.; Wang, P. Role of Thrombosis in Osteonecrosis. Curr. Hematol. Rep. 2003, 2, 417–422.
- van Giezen, J.J.; Jansen, J.W. Correlation of in Vitro and in Vivo Decreased Fibrinolytic Activity Caused by Dexamethasone. Ann. N. Y. Acad. Sci. 1992, 667, 199–201.
- Kerachian, M.A.; Séguin, C.; Harvey, E.J. Glucocorticoids in Osteonecrosis of the Femoral Head: A New Understanding of the Mechanisms of Action. J. Steroid Biochem. Mol. Biol. 2009, 114, 121–128.
- Glueck, C.J.; Fontaine, R.N.; Gruppo, R.; Stroop, D.; Sieve-Smith, L.; Tracy, T.; Wang, P. The Plasminogen Activator Inhibitor-1 Gene, Hypofibrinolysis, and Osteonecrosis. Clin. Orthop. Relat. Res. 1999, 366, 133–146.
- Pósán, E.; Hársfalvi, J.; Szepesi, K.; Gáspár, L.; Batár, P.; Udvardy, M. Increased Platelet Activation and Decreased Fibrinolysis in the Pathogenesis of Aseptic Necrosis of the Femoral Head. Platelets 1998, 9, 233–235.
- Zhang, Q.; Lv, J.; Jin, L. Role of Coagulopathy in Glucocorticoid-Induced Osteonecrosis of the Femoral Head. J. Int. Med. Res. 2018, 46, 2141–2148.
- Pacinella, G.; Ciaccio, A.M.; Tuttolomondo, A. Endothelial Dysfunction and Chronic Inflammation: The Cornerstones of Vascular Alterations in Age-Related Diseases. Int. J. Mol. Sci. 2022, 23, 15722.
- Esper, R.J.; Nordaby, R.A.; Vilariño, J.O.; Paragano, A.; Cacharrón, J.L.; Machado, R.A. Endothelial Dysfunction: A Comprehensive Appraisal. Cardiovasc. Diabetol. 2006, 5, 4.
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 2019, 124, 315–327.
- Fan, T.; Song, Y.-J.; Liu, X.-L. Adenocarcinoma of the Lung with Concomitant ALK Fusion Gene and EGFR Gene Mutation: A Case Report and Literature Review. Mol. Clin. Oncol. 2016, 4, 203–205.
- Parikh, S.; Gomez, O.; Davis, T.; Lyon, Z.; Corces, A. Avascular Necrosis as a Sequela of COVID-19: A Case Series. Cureus 2023, 15, e35368.
- Agarwala, S.R.; Vijayvargiya, M.; Sawant, T. Secondary Osteonecrosis of the Knee as a Part of Long COVID-19 Syndrome: A Case Series. BMJ Case Rep. 2022, 15, e248583.
- Hassan, A.A.A.; Khalifa, A.A. Femoral Head Avascular Necrosis in COVID-19 Survivors: A Systematic Review. Rheumatol. Int. 2023, 43, 1583–1595.
- Hong, Y.-C.; Zhong, H.-M.; Lin, T.; Shi, J.-B. Comparison of Core Decompression and Conservative Treatment for Avascular Necrosis of Femoral Head at Early Stage: A Meta-Analysis. Int. J. Clin. Exp. Med. 2015, 8, 5207–5216.
- Tan, Y.; He, H.; Wan, Z.; Qin, J.; Wen, Y.; Pan, Z.; Wang, H.; Chen, L. Study on the Outcome of Patients with Aseptic Femoral Head Necrosis Treated with Percutaneous Multiple Small-Diameter Drilling Core Decompression: A Retrospective Cohort Study Based on Magnetic Resonance Imaging and Equivalent Sphere Model Analysis. J. Orthop. Surg. Res. 2020, 15, 264.
- Hong, Y.-C.; Luo, R.-B.; Lin, T.; Zhong, H.-M.; Shi, J.-B. Efficacy of Alendronate for Preventing Collapse of Femoral Head in Adult Patients with Nontraumatic Osteonecrosis. BioMed Res. Int. 2014, 2014, 716538.
- Ma, Y.; Wang, T.; Liao, J.; Gu, H.; Lin, X.; Jiang, Q.; Bulsara, M.K.; Zheng, M.; Zheng, Q. Efficacy of Autologous Bone Marrow Buffy Coat Grafting Combined with Core Decompression in Patients with Avascular Necrosis of Femoral Head: A Prospective, Double-Blinded, Randomized, Controlled Study. Stem Cell Res. Ther. 2014, 5, 115.
- Tang, H.-Y.; Zhao, Y.; Li, Y.-Z.; Wang, T.-S. Effectiveness of Extracorporeal Shock Wave Monotherapy for Avascular Necrosis of Femoral Head: A Systematic Review Protocol of Randomized Controlled Trial. Medicine 2019, 98, e15119.
- Tu, Y.; Chen, Z.; Lineaweaver, W.C.; Zhang, F. Different Recipient Vessels for Free Microsurgical Fibula Flaps in the Treatment of Avascular Necrosis of the Femoral Head: A Systematic Review and Meta-Analysis. Ann. Plast. Surg. 2017, 79, 583–589.