Uric acid is the final product of purine metabolism and is converted to allantoin in most mammals via the uricase enzyme. The accumulation of loss of function mutations in the uricase gene rendered hominoids (apes and humans) to have higher urate concentrations compared to other mammals. The loss of human uricase activity may have allowed humans to survive environmental stressors, evolution bottlenecks, and life-threatening pathogens. While high urate levels may contribute to developing gout and cardiometabolic disorders such as hypertension and insulin resistance, low urate levels may increase the risk for neurodegenerative diseases. The double-edged sword effect of uric acid has resurrected a growing interest in urate’s antioxidant role and the uricase enzyme’s role in modulating the risk of obesity.
1. Background
Uric acid is the final product of purine metabolism in humans. Serum urate levels vary significantly between different species. Particularly, humans have the highest urate levels compared to all other mammals. Markedly high uric acid levels in humans have been caused by the loss of the uricase enzymatic activity during the Neogene period [
1,
2].
In humans, two-thirds of uric acid is excreted through the kidney and one-third through the gastrointestinal tract [
3]. While humans do not produce uricase per se, a growing body of literature has suggested that the gut microbiome, a primary source of uricase, may have a role in compensating for the loss of the
uricase gene [
4,
5,
6,
7]. Increased urate production or underexcretion of uric acid can increase serum urate levels beyond the solubility threshold, accelerating the formation of monosodium urate (MSU) crystals in and around the joints.
The loss of the uricase enzyme in humans and the extensive reabsorption of uric acid suggest that retaining uric acid is essential to human health; hence, evolutionary physiology has treated uric acid not as a harmful waste. Also, this high conservation of uric acid has rendered humans susceptible to uric acid changes induced by diet and their propensity to develop gout spontaneously. Despite the well-established relationship between high uric acid levels and gout, researchers have been actively trying to elucidate the health benefits of uric acid, its role in adaptation to different environments, and potentially inform the development of less immunogenic uricase-based gout therapy [
17].
Proposed theories about the role of uric acid have been previously described [
1]. For example, growing evidence suggests that uric acid is a powerful antioxidant, accounting for roughly 50–60% of those naturally occurring in humans [
18]. This naturally occurring protection may have elongated the life expectancy of hominoids (apes and humans). While two-thirds of uric acid levels are because of endogenous sources, one-third of uric acid is the result of external factors, mainly diet and certain lifestyle factors.
2. Genetic Correlation between Urate Levels and Cardiometabolic Traits
Developing hyperuricemia or gout is multifactorial, including genetics, dietary habits, and other lifestyle factors [
20]. Multiple studies have demonstrated that certain risk factors, such as high body mass index (BMI), male sex, advanced age, and increased consumption of meat, alcohol, and high-fructose corn syrup, could increase uric acid levels, increasing the risk of gout [
3]. Other factors, such as genetic polymorphisms across major uric acid transporters, have been linked to uric acid levels and the risk of developing gout. For instance, genetic polymorphisms identified through genome-wide association studies (GWAS), including
SLC2A9,
SLC16A9,
SLC17A1,
SLC22A11,
SLC22A12,
ABCG2,
PDZK1,
GCKR,
LRRC16A1,
HNF4A/G, and
INHBC are some of the major determinants in uric acid levels and modulators of gout risk [
23,
24].
Serum urate levels are highly heritable, with an estimate of 30–60%. Despite the modest degree of heritability of urate levels, the variability explained in serum urate levels by constructing genetic models remains limited to up to 17% using SNPs at 183 loci [
28]. In contrast, non-genetic models could explain a higher proportion of the variability in serum urate and precisely predict the risk of gout compared to traditional genetic risk models. In a study using UK Biobank data, the genetic risk model was a significantly weaker predictor (area under the receiver operating characteristic curve (AUROC) = 0.67) than the demographic model (age and sex) (AUROC = 0.84) [
28].
Multiple major epidemiological studies repeatedly showed a strong association between serum urate levels with multiple cardiometabolic traits and other cardiovascular risk factors [
31,
32,
33]. Using the National Health and Nutrition Examination Survey, the conditional probability of obesity, given the individual has hyperuricemia, is between 0.4 and 0.5 [
34]. Similarly, the prevalence of hypertension, diabetes, and chronic kidney disease proportionally increases as uric acid levels increase; the same risk factors are more common in patients with gout than those without gout [
35]. With a robust genetic underpinning, the genetic correlation between urate levels and complex diseases has been evaluated [
28,
30].
3. Uricase Activity: Fructose Metabolism and Energy Storage
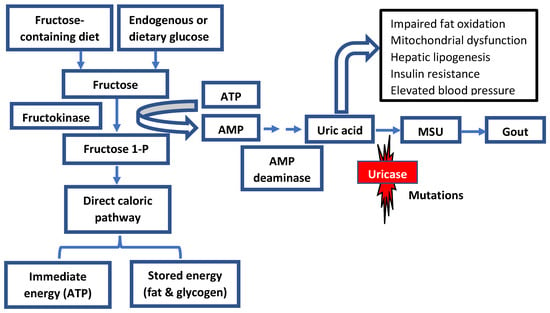
The slow decline in uricase activity has been linked to climate and vegetation conditions associated with food scarcity and famine periods. It is hypothesized that the progressive decrease in uricase activity enabled our ancestors to readily accumulate and store fat via the metabolism of fructose to fructose-1-phosphate from fruits in anticipation of significant climate changes or droughts [
15]. Additionally, increased uric acid levels because of declining uricase activity could have amplified the effect of fructose on energy and fat storage (
Figure 2).
Figure 2. Role of fructose metabolism in fat and energy storage in humans. The loss of uricase activity could amplify the role of uric acid in response to fructose. Both fructose and uric acid would have led to the activation of the biological response to starvation, leading to increased fat and energy storages. Fructose-1-P: Fructose-1- Phosphate; AMP: Adenosine monophosphate; MSU: Monosodium urate.
While high serum urate levels may enhance the development of metabolic syndrome, uric acid could further potentiate the metabolism of fructose by increasing the expression of fructokinase, which in turn can increase the formation of fructose-1-phosphate and the formation of energy storages such as glycogen and triglycerides (
Figure 2) [
37].
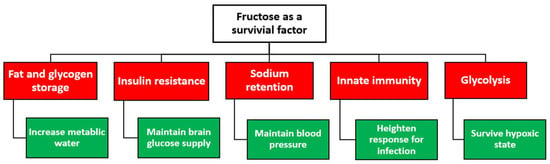
Figure 3. The proposed role of fructose as a survival factor in response to starvation in early hominoids.
Evidence from animal research has demonstrated excessive consumption of fructose sources in preparation for food, oxygen, and water scarcity. Fructose ingestion has been shown to conserve water by stimulating vasopressin, which reduces water loss through the kidney and stimulates fat and glycogen production as a source of metabolic water [
38].
Natural selection for losing uricase activity has enabled hominoids to retain high uric acid levels, which may have enhanced fructose metabolism and increased fat stores. Combined, increased uric acid levels because of the lost uricase activity might have provided a survival advantage during those critical historical times. These harsh environments may have ushered in the introduction of biological mutations that improved survival in the setting of starvation. The same notion may be responsible for the current obesity epidemic, where food is plentiful and global fructose consumption is rising [
40]. These observations may collectively support the thrifty genes hypothesis: individuals who could easily store extra energy would have had an evolutionary advantage during famines and partly explain the global rise in obesity and metabolic syndrome [
41,
42,
43].
4. Regulating Blood Pressure: The Role of Salt and Uric Acid
A direct relationship between salt intake and blood pressure (BP) has been well established. Excessive sodium intake has been shown to increase blood pressure and the onset of hypertension. Increased sodium consumption is associated with increased water retention, peripheral vascular resistance, oxidative stress, and microvascular remodeling [
44]. Following this growing evidence, salt restriction has become a pillar in hypertension management and patient counseling. However, the BP response to changing salt intake displays marked inter-individual variability. Therefore, salt sensitivity could be viewed as a continuous parameter at the population level. Inter-individual variability in the BP response to salt modifications could be compounded by
While being a naturally occurring antioxidant, uric acid has been strongly implicated in developing the hypertension epidemic and maintaining blood pressure during the early hominoids. Fossil evidence suggests that early hominoids heavily relied on fruits for their dietary needs. With a fruit-based diet, the sodium intake during the Miocene period was expected to be very low to maintain blood pressure. To evaluate this hypothesis, mild hyperuricemia was induced in rats by inhibiting uricase, recreating the pre-historic conditions by feeding the rats a low-sodium diet. Compared with hyperuricemic rats, normal rats receiving a low-sodium diet experienced either no change or a drop in systolic blood pressure. In contrast, systolic blood pressure increased in hyperuricemic rats, proportional to uric acid levels, and could be mitigated using allopurinol [
45,
46]. Therefore, it was presumed that increased uric acid levels may have maintained blood pressure during low sodium intake.
5. Hyperuricemia and Hypertension: A Cause or Effect?
Inflammation remains the principal suspect linking gout and cardiovascular diseases. Mechanisms whereby serum urate levels could contribute to the development of hypertension are partly related to the uric acid’s primary effect on the kidney. These mechanisms include activating the renin–angiotensin–aldosterone system (RAAS) and the disposition of urate crystals in the urinary lumen. Additional evidence suggests that elevated serum uric acid can decrease nitric oxide production, causing endothelial injury and dysfunction. It is well established that monosodium urate crystals can activate the NLRP3 inflammasome cascade and the production of inflammatory cytokines such as IL-1 beta and IL-8 [
47]. Although uric acid is predominantly eliminated through the kidney, new evidence has reported that urate crystals can deposit in the coronary artery among gout patients [
48]. The deposition of urate crystals in the aorta and main arteries was associated with a higher coronary calcium score and can trigger an inflammatory response like the observed response in the kidney. Beyond the joints, these observations support the hypothesis that high serum urate levels could deposit into soft tissues and extraarticular regions, which can then become pro-oxidants, increasing the inflammatory burden and the development of the multiple comorbidities observed in patients with gout or hyperuricemia [
49].
Hypertension is an epidemic affecting more than 30% of adults worldwide [
50]. While genetics may contribute to the development of hypertension, the contribution has been limited, suggesting that the external environment plays a significant role in developing hypertension compared with genetics. Moreover, a growing body of evidence has repeatedly reported on the association between elevated urate levels (hyperuricemia) and hypertension. For example, a cross-sectional study determined that each 1 mg/dL increase in serum urate contributes to a 20% increased prevalence of hypertension in a general population not treated with hyperuricemia and hypertension [
51].
Hyperuricemia is an independent risk factor for multiple cardiovascular disease risk factors, which can, collectively, elevate the risk of cerebrovascular events. Nonetheless, the direct role of uric acid levels in developing stroke events is inconclusive [
53,
54].
The incidence and prevalence of hypertension and obesity were minimal in specific populations until Westernization and the adoption of a Western diet [
57,
58]. Additionally, the immigration of select population subgroups, Filipinos and Japanese, has been linked to earlier gout onset, higher gout and hypertension rates, and higher mean urate levels than their native home residents [
25,
58,
59,
60,
61,
62]. These observations support the hypothesis that dietary changes from a traditional diet to a Westernized diet enriched with fatty meats may be responsible for the increased prevalence of gout, high serum urate levels, and increased incidence of hypertension and diabetes.
6. Uric Acid and Neurodegenerative Diseases: The Antioxidant Hypothesis
The old hypothesis that the chemical structure of uric acid could mimic specific brain stimulants, such as caffeine or methylxanthine, may have provided selective advantages to early hominoids [
63]. To this hypothesis, the loss of uricase activity in early hominoids has caused an increase in uric acid levels in the brain, which could have given rise to quantitative and qualitative intelligence among early hominoids. Indeed, limited studies have shown that urate plasma levels may be associated with higher intelligence [
63,
64,
65].
Multiple epidemiological studies have examined the association between serum urate levels, gout diagnosis, and brain-related diseases [
66,
67]. Specifically, the associations between urate levels or gout and Alzheimer’s disease (AD) and Parkinson’s disease (PD) have been a focal point in recent studies [
68,
69,
70]. However, the results of these studies have been conflicting or showing no association [
71,
72]. Partly, these inconclusive findings have been attributed to the study design, not accounting for uric acid level confounders, and the varying degrees of disease progression among study subjects [
73]. While the transport of uric acid into the brain remains elusive, uric acid is produced in the human brain [
69]. Urate production in the brain has paved a path for a great deal of research into whether uric acid could exert antioxidant activity, protecting the brain from developing neurodegenerative diseases by mitigating the burden of oxidative stress sequelae. Uric acid can function as an antioxidant and block peroxynitrite, which could be beneficial in slowing the progression of PD and multiple sclerosis (MS) [
74,
75,
76]. Lower uric acid levels have been linked with a greater risk of developing PD, the severity of motor features, and faster progression of both motor and non-motor features [
69]. This observation has led to the hypothesis that raising uric acid may provide a neuroprotective effect in PD patients [
77].
To assess the causality between uric acid and PD, a Mendelian randomization approach was used to systematically evaluate the inherent risk of lower urate levels and disability requiring dopaminergic treatment in early PD patients. Genotyping 808 patients for three loci across the
SLC2A9 (rs6855911, rs7442295, and rs16890979), cumulative scores of the risk alleles were created based on the total number of minor alleles across the three loci [
78]. Serum urate levels were 0.69 mg/dL lower among individuals with ≥4
SLC2A9 minor alleles versus those with ≤2 minor alleles [
78]. A 5% increased risk of PD progression with a 0.5 mg/dL decrease in serum urate was observed. However, the hazard ratio for the progression to disability requiring dopaminergic treatment, though increased with the
SLC2A9 score, was not statistically significant (
p = 0.056) [
78].
The role of uric acid levels in mild cognitive impairment (MCI), a common diagnosis that precedes the development of clinical AD, has been extensively investigated. While asymptomatic hyperuricemia can go undiagnosed and rarely treated, exploring the association between hyperuricemia and neurodegenerative diseases may be confounded by recall bias, disease misdiagnosis, and secondary causes of hyperuricemia. Nonetheless, patients with gout are considered to have the highest uric acid burden and represent the extreme phenotype of hyperuricemia. Therefore, the association between gout diagnosis and the risk of neurodegenerative diseases has been of significant interest in neurodegenerative disease research.
7. Hyperuricemia and Innate Immune System: Acquired Protection?
Selective advantages to developing hyperuricemia in distinct population groups may have been driven by natural selection. This natural selection advantage may partly explain the inherently increased predisposition for hyperuricemia or gout among select population groups, especially when challenged by unprecedented environmental threats or culturally altering events. Besides being a naturally occurring antioxidant, urate levels may have enabled humans to survive crucial environmental stressors and pathogenic threats. When crystallized, urate may trigger the innate immune response. For this reason, monosodium urate crystals are considered a critical natural endogenous immune response trigger. With infectious diseases being one of the strongest drivers of natural selection, it is presumed that genes associated with raising urate may have been selected during major depopulation events. This theory may partly explain the genetic predisposition for hyperuricemia/gout and the higher mean uric acid levels in the Pacific and Oceania regions compared with whites and the Western world [
25,
87,
88,
89,
90].
Historically, exposure to pathogenic or parasitic infections such as malaria has pressured the human genome of distinct populations to develop disorders like sickle-cell disease and glucose-6-phosphate dehydrogenase deficiency as a survival switch. Therefore, it is plausible that the inherent risk of hyperuricemia could be considered another adaptation to protect against malaria or other pathogens that threatened our ancestors. Urate levels released from erythrocytosis because of malaria infection have been shown to activate the host’s inflammatory response, facilitating the detection of the parasite [
87].
8. Uric Acid: A Therapeutic Target or Disease Bystander?
Uric acid levels are the strongest predictor of developing gout via the deposition of monosodium urate crystals in and around the joints. The chronic suppression of uric acid levels is the hallmark of reducing the MSU crystal burden and preventing future gout flares. While different treatment modalities have been developed to lower serum urate, most treatments are focused on inhibiting uric acid production. In the advanced forms of gout, the recombinant form of uricase has been used to reduce the tophi burden or if the patient is not responsive to available treatments.
While numerous experimental and clinical studies support the role of uric acid as an independent cardiovascular risk factor, a handful of studies also suggested that lowering serum urate may improve blood pressure. These studies included pre-hypertensive obese [
95], hypertensive adolescents [
96], hypertensive children on an angiotensin-converting enzyme inhibitor [
97], and adults with asymptomatic hyperuricemia [
98,
99].
A growing body of evidence also suggests that urate-lowering therapy does not affect blood pressure. A crossover single-center study enrolled ninety-nine participants randomized to 300 mg of allopurinol or placebo over four weeks [
100]. There was no difference in the change in blood pressure between the groups; however, the allopurinol group had a decrease in uric acid and improved endothelial function, estimated as flow-mediated dilation [
100]. What is noteworthy is that the study participants were relatively young (28 years old), had relatively normal systolic blood pressure (123.6 mm Hg), and had a relatively normal uric acid level at baseline (5.9 mg/dL) [
100].
With gout and hyperuricemia being associated with increased sterile inflammation, earlier reports suggested that the use of allopurinol in patients with hyperuricemia can significantly reduce inflammatory biomarkers [
98]. A recent study showed that urate-lowering therapy, mainly allopurinol, was significantly associated with lower hs-CRP, LDL, and total cholesterol levels in patients with gout compared with those not receiving urate-lowering therapy [
102]. These results support that patients optimally treated for gout can garner added benefits above and beyond reduced uric acid burden. Although urate-lowering therapy was not associated with lowering blood pressure, a high chronic kidney disease diagnosis in patients receiving allopurinol was observed [
102]. Overall, the study suggested that uric acid may be a remediable risk factor to lower the atherogenic disease burden in patients with gout [
102]
9. Summary
The loss of activity mutations in the uricase gene may have been protective in situations of famine and food scarcity; thus, it rapidly took over the ancestral population, likely driven by harsh environmental factors during the Miocene era. In modern times, this adaptation is possibly leading to a state of hyperuricemia, increasing the risk of gout and other cardiometabolic disorders. Today, all humans are considered uricase knockouts. Loss of the uricase enzyme resulted in the inability to regulate uric acid effectively. The adaptation to famine and food scarcity in early hominoids dictated crucial changes to obtain and conserve energy for survival. In contrast to animal experiments and observational studies, targeting uric acid levels for therapeutic purposes beyond the purpose of gout remains conflicting.
As hyperuricemia and gout animal models continue to be instrumental in advancing the field of gout and uric acid metabolism, it is equally important to recognize that animal models are inherently predisposed to eliminating uric acid. Therefore, the effects of raising uric acid levels in animal models may not reflect the outcomes seen in human studies. Additionally, the sex effect on uric acid levels reinforces the need for adequate and robust analyses by sex.
While genetics play a significant role in developing hyperuricemia or gout, it is equally important to recognize that the same genetics are therapeutic targets to modify uric acid metabolism. Genetic polymorphisms have been implicated in the racial difference in gout prevalence and may also contribute to the heterogeneity in response to urate-lowering or urate-elevating therapies. Therefore, genetic investigations into uric acid metabolism in clinical trials need to be considered in designing the study. Finally, as described earlier, uric acid levels are the culmination of endogenous cellular processes and external dietary sources; therefore, accounting for dietary and lifestyle habits in uric acid levels may minimize the effect of study confounders.
This entry is adapted from the peer-reviewed paper 10.3390/jpm13091409