Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Human cytomegalovirus (HCMV) is a herpesvirus capable of establishing a lifelong persistence in the host through a chronic state of infection and remains an essential global concern due to its distinct life cycle, mutations, and latency. It represents a life-threatening pathogen for immunocompromised patients, such as solid organ transplanted patients, HIV-positive individuals, and hematopoietic stem cell recipients. Multiple antiviral approaches are available and administered in order to prevent or manage viral infections in the early stages.
- HCMV
- treatment
- antiviral drugs
1. Introduction
Biological features. Human cytomegalovirus (HCMV), also known as human herpesvirus 5 (HHV-5), is the prototype member of the Betaherpesvirinae and the largest member of the virus family Herpesviridae [1]. It is a ubiquitous virus that infects almost all humans at some time in their lives. The virus was first isolated by three different groups of investigators: Rowe and colleagues, Weller and colleagues, and Smith simultaneously in 1956 [2]. The genome is a linear, double-stranded DNA molecule with a 236 ± 1.9 kbp size divided into a unique long (UL) and a unique short (US) region, both of which are flanked by terminal and internal repeats [3]. In detail, it contains more than 751 translated open reading frames (ORFs) of which 282 translationally active viral transcripts, 4 major long non-coding RNAs (lncRNAs) (RNA1.2, RNA2.7, RNA4.9, and RNA5.0) and at least 16 pre-miRNAs and 26 mature miRNAs [4]. Despite enclosing a much larger genome, the size of the HCMV capsid is similar to that of other herpesviruses (130 nm), structured as an icosahedral ordered nucleocapsid with triangulation number (T) = 16 and composed of 162 capsomers, divided into two distinct morphological units, 12 pentamers, and 150 hexamers [5][6][7]. Externally to the capsid, the tegument is located, which is generally thought to be unstructured and amorphous in nature. However, some structuring is observed with the binding of tegument proteins to the protein capsid. The tegument proteins are usually phosphorylated and comprise more than half of the total proteins found within infectious virions [8][9]. Finally, a lipidic bilayer envelope membrane, containing eighteen proteins including four viral G protein-coupled receptors (pUL33, pUL78, pUS27, and pUS28), covers the tegument and HCMV nucleocapsid. This envelope is similar in structure and composition to host cell membranes [10][11][12].
Life cycle and pathogenesis. Similar to other herpesviruses, HCMV establishes a persistent infection, remaining silent in the host and undergoing productive reactivation cycles that contribute to its efficient transmission. HCMV is able to replicate in a wide variety of cells (epithelial and mucosal cells, smooth muscle cells, fibroblasts, macrophages, dendritic cells, hepatocytes, and endothelial cells), thereby allowing for systemic spread in the human body and among host [13].
HCMV enters human cells either through direct fusion or an endocytic pathway. The virus attaches to the cell via interactions between viral anti-receptors (gH/gL/pUL128L pentamer complex, and gH/gL/gO trimer complex) and specific surface cell receptors (PDGFRα, Nrp2, and OR14I1), followed by gB activation to fuse the virus envelope with the cellular membrane [14]. Nucleocapsids are released into the cytoplasm and subsequently translocated to the nucleus, where they release viral DNA. HCMV genes are expressed in a sequential cascade, with temporal phases designated immediate-early (IE), early, and late. The major IE genes (MIE) UL123 and UL122 (IE1/IE2) are the first genes to be coded and, together with cellular host factors, coordinate the next level of gene expression (early (E) genes) involved in viral replication [15][16]. Typical early viral proteins include the DNA polymerase (pUL54), phosphotransferase (pUL97), and terminase components (pUL51, pUL52, pUL56, pUL77, pUL89, pUL93, and pUL104) [17]. Finally, HCMV encodes distinct categories of late genes, commonly referred to as leaky late (γ1) and true late (γ2): the former are expressed independently of viral DNA synthesis, while the latter are not expressed at all when viral DNA synthesis is blocked by specific inhibitors [18]. True late genes generally encode structural proteins required for the assembly of new virions, such as pUL77, pUL93 pUL115, pp28, and pp150 [19].
After DNA replication, the following steps are the encapsulation of the replicated viral DNA as capsids, which are then transported from the nucleus to the cytoplasm and coiling in the intermediate compartment of the endoplasmic reticulum (ER)-Golgi. This is then followed by a final complex two-stage envelopment and egress process that leads to virion release by exocytosis at the plasma membrane [20].
This lytic infection program leads to the release of infectious virions and can occur in an array of cells and tissues, while alternatively, in some cell types (CD14+ monocytes and their CD34+ progenitor), the virus can enter a latent life cycle that is associated with a much more limited viral transcription program and a lack of virion production [21].
Epidemiology and transmission routes. HCMV is a global herpesvirus highly prevalent worldwide with a prevalence of about 100% in both Africa and Asia and 45.6–95.7% in Europe and North America [22]. The heterogeneous HCMV seroprevalence appears to be related to race, ethnicity, socioeconomic status, and education level [23].
Treatment. The antiviral approach for the treatment of HCMV infections relies on different drugs, such as inhibitors of viral DNA polymerase, nucleoside and nucleotide analogs, pyrophosphate analogs, and terminase inhibitors [24][25][26]. Currently, various strategies such as preemptive therapy, antiviral prophylaxis, hybrid approaches (continuous surveillance after prophylaxis for HCMV viremia with preemptive therapy), and HCMV-specific immunity-guided approaches could be used for the effective control of HCMV infection in transplanted patients [27]. However, antiviral prophylaxis and preemptive therapy are the most commonly used strategies worldwide. In antiviral prophylaxis, antiviral drugs are routinely administered to all transplant recipients at risk for HCMV disease, typically for 3 months or more immediately after transplantation, while in a preemptive therapy strategy, HCMV DNAemia is measured according to a predetermined time schedule, and antiviral drugs are administered to those transplant recipients in whom the HCMV DNA level reaches alert thresholds while the infection is still asymptomatic. Under such conditions, only a restricted cohort of patients is treated for a reduced period. [28][29]. The antiviral prophylaxis resulted in superior control of HCMV infection and prolonged time to HCMV disease in transplanted recipients without an increased risk of opportunistic infections, graft loss, drug-related adverse effects, the development of drug resistance, and mortality.
2. Antiviral Approach to Human Cytomegalovirus Infection
Ganciclovir (C9H13N5O4). Ganciclovir (GCV) is a synthetic nucleoside analog of 2′-deoxy-guanosin, whose primary mechanism is the inhibition of the viral DNA polymerase. The drug is primarily converted intracellularly to Ganciclovir 5′-monophosphate by a viral kinase encoded by the HCMV gene UL97 during infection. Subsequently, cellular kinases (deoxyguanosine kinase, guanylate kinase, and phosphoglycerate kinase) catalyze the formation of Ganciclovir diphosphate and Ganciclovir triphosphate. This latter is present in 10-fold greater concentrations in HCMV-infected cells than in uninfected cells [30][31]. The triphosphate form of Ganciclovir competitively inhibits the viral DNA polymerase, being inserted into the replicative HCMV genome, thus leading to premature termination and halting further viral DNA synthesis. This action disrupts the spread of HCMV and reduces the viral load.
Ganciclovir was the first antiviral agent approved for the treatment of HCMV infection, and it is mostly administered as an intravenous formulation due to its low oral bioavailability. To overcome this limit, the prodrug Valganciclovir (VGCV), a GCV valine ester, was developed. It showed higher absorption after oral administration (60.9% compared to 5.6% bioavailability of GCV), and it was rapidly metabolized to active form GCV [32]. GCV dosage in transplanted patients relies on various factors, including the type of transplanted organ, the patient’s weight, renal function, and the severity of the HCMV infection. Usually, the induction therapy dosage is between 1.25 mg/Kg and 5.0 mg/Kg every 12/24 h for 7 to 14 days, depending on patients’ age and weight, while the maintenance dosage starts from 0.625 mg/Kg every 24 h. According to the recipient’s clinical condition, the maintenance therapy phase can be performed via oral or intravenous administration. For induction and maintenance therapy, Valganciclovir dosage is between 450 and 900 mg once or twice every day, according to renal function. Meanwhile, prophylactic dosage to prevent HCMV disease relies on oral administration of 900 mg once daily [33][34]. The most common side effect associated with both Ganciclovir and Valganciclovir is leukopenia. Moreover, a recent systemic review of 102 studies (25 human and 77 animal) assessing the long-term effects in subjects receiving a prophylactic dose of Ganciclovir observed a correlation with spermatic toxic effects in those patients [35]. Despite the effectiveness of Ganciclovir and Valganciclovir, HCMV resistance can occur due to mutations targeted to the viral kinase or DNA polymerase genes. Mutations in the viral UL97 gene confer resistance to Ganciclovir, whereas mutations in the UL54 DNA polymerase gene are typically associated with high-level resistance to Ganciclovir and cross-resistance to Cidofovir and Foscarnet. Common mechanisms of HCMV resistance to Ganciclovir have been described predominantly with the UL97 mutation and occurred at codons 460–607, with mutations at codons 460 and 520 resulting in at least a five-fold increase in IC50 [36].
Cidofovir (C8H14N3O6P). Cidofovir (CDV) is a cytosine monophosphonate nucleotide analog cytosine with potent broad-spectrum antiviral activity. Unlike Aciclovir and other nucleoside analogs, which require monophosphorylation by viral kinases for activation, Cidofovir already carries a phosphonate group and does not require viral enzymes for conversion to Cidofovir diphosphate, the active antiviral compound [37]. It undergoes two stages of phosphorylation via monophosphate kinase and pyruvate kinase in order to form the active form triphosphate. Cidofovir triphosphate acts as a competitive inhibitor of HCMV DNA polymerase, encoded by the UL54 gene, preventing the incorporation of deoxycytidine triphosphate (dCTP) into growing viral DNA [38]. The active form of the drug exhibits a 25- to 50-fold greater affinity for the viral DNA polymerase, compared with the cellular DNA polymerase, thereby selectively inhibiting viral replication [39]. However, since Cidofovir is a nonobligate chain terminator, the incorporation of CDV diphosphate into the nascent strand of DNA does not necessarily result in termination [37].
Cidofovir is administered via intravenous infusion in conjunction with oral probenecid to reduce nephrotoxicity. The induction therapy relies on 5 mg/Kg once a week for two weeks, while maintenance therapy should be performed at the dosage abovementioned once every two weeks. Cidofovir was not recommended for prophylaxis. It is reserved for the treatment of Ganciclovir-resistant or refractory HCMV disease, and its use is complicated by a high rate of nephrotoxicity due to Cidofovir’s high affinity for the organic anion transporter in the convoluted proximal tubules and is responsible for cell necrosis proximal tubes [40]. UL54 DNA polymerase mutations typically add to pre-existing UL97 mutations after prolonged Ganciclovir therapy and increase the overall level of drug resistance [41].
A lipid-conjugated Cidofovir-derived prodrug, Brincidofovir (C27H52N3O7P), showed higher antiviral activity in vitro compared to Cidofovir and also in preventing HCMV reactivation. However, it failed all clinical trials due to adverse side effects, and it is not currently used [42].
Foscarnet (CH3O5P). Foscarnet (FOS) is a pyrophosphate analog, an oxyanion of inorganic phosphorous, that functions as a noncompetitive inhibitor of the herpesvirus DNA polymerase of all HHVs, including the most Ganciclovir-resistant HCMV isolate and Acyclovir-resistant HSV and VZV strains [39][43]. This analog acts like the pyrophosphate molecule by selectively and reversibly binding to the binding site on the HCMV DNA polymerase and inhibiting further DNA chain elongation. The role of the DNA polymerase enzyme is to cleave the pyrophosphate molecule from the DNA chain to add further nucleotides to the growing chain. Foscarnet binds and blocks that cleaving process. Although Foscarnet has selectivity for the viral DNA polymerase, it can also inhibit human DNA polymerase in much higher drug concentrations [44]. Along with Cidofovir, Foscarnet is considered a second-line agent reserved for the treatment of resistant and refractory HCMV. Foscarnet is not an orally administered drug due to low bioavailability and its inclination to be deposited within bone and cartilage. Instead, the most common route is intravenous administration, and the dosage and rate of administration are determined based on the patient’s age and weight and the specific viral infection (HCMV versus HSV or VZV). Usually, for Ganciclovir-resistant HCMV and AIDS-associated HCMV retinitis, the induction therapy is 90 mg/Kg intravenous administration every 12 h for 2 to 3 weeks or, alternately, 60 mg/Kg every 8 h for 2 to 3 weeks. Meanwhile, maintenance treatment is performed with 90 mg/Kg every 24 h or 120 mg/Kg every 24 h [30][44]. Foscarnet-associated nephrotoxicity affects 30–50% of patients after long-term use due to the deposition of drug crystals in the glomerular capillary lumen. Myelosuppression, mucosal ulcerations, and electrolyte disturbances such as hypocalcemia, hypomagnesemia, and hypophosphatemia are also common [45][46].
Foscarnet resistance mutations encountered in clinical practice are clustered in the domains of the DNA polymerase structure designated in amino terminal 2 (residues 555–600), the palm, and the finger (residues 696–981) and typically confer 3- to 5-fold decreases in antiviral susceptibility with variable low-grade cross-resistance to Ganciclovir/Valganciclovir and sometimes to Cidofovir as well [47].
Letermovir (C29H28F4N4O4). Letermovir (LMV) is a 3,4-dihydro-quinozoline that acts through inhibition of the viral terminase enzyme complex (pUL51, pUL56, and pUL89) used by HCMV in the terminal replication life cycle stage of viral DNA processing and packaging [48]. Letermovir is highly specific for HCMV, as it has no activity against other herpesviruses or any other virus, and it is one of the most potent anti-HCMV agents identified to date, with reports illustrating up to 1000 times the potency of Ganciclovir [49]. It was approved in 2017 for HCMV prophylaxis in HCMV-seropositive adult hematopoietic cell transplant (HCT) recipients and has been widely adopted in this population, but it is currently not approved for any clinical indication in solid organ transplant recipients [50][51]. The approved dosage of Letermovir is 480 mg (240 mg if co-administered with cyclosporine) once daily. Actually, it is recommended to start Letermovir at this dose in HCMV-seropositive adult patients who received an allo-HCT between days 0 and 28 and continue until day 100 post-transplantation. The route of administration is oral, and no dose adjustment is required for renal or liver impairment [52]. Letermovir is a generally well-tolerated drug, and the most commonly reported side events during clinical trials were diarrhea, nausea, and vomiting. Differently from the antiviral drugs abovementioned, it does not appear to have significant nephrotoxicity or myelosuppressive effects. Only one case of self-limiting hepatitis thought to be due to Letermovir has been reported [53].
HCMV resistance to Letermovir has emerged in both experimental and clinical settings. Mutations conferring resistance to Letermovir are most commonly mapped to UL56 (particularly codons 231–369, e.g., V236M, L241P, and R369S). Hoverer, rarer mutations of UL51 and UL89 have been implicated in the emergence of resistance. They have been described when LMV was administered as salvage therapy for drug-resistant or refractory HCMV infections and when used as a primary or secondary prophylaxis [54][55].
Maribavir (C15H19Cl2N3O4). Maribavir is a benzimidazole l-riboside antiviral compound that inhibits UL97 protein kinase and its natural substrates, thereby inhibiting HCMV DNA replication, encapsidation, and nuclear egress. This drug acts by blocking the phosphorylation of several downstream viral proteins, including UL44, thus inhibiting HCMV replication. In vitro, Maribavir is effective for HCMV and Epstein–Barr viruses but has no activity against Herpex Simplex virus 1 (HSV-1), HSV-2, Varicella Zoster virus, Human Herpesvirus 6 (HHV-6), or HHV-8 [56]. Unlike Valganciclovir/Ganciclovir, Maribavir targets a different location on pUL97 and does not require intracellular processing by pUL97 protein kinase [57].
It received U.S. Food and Drug Administration (FDA) approval in November 2021 for the treatment of adult and pediatric patients (12 years of age and older, weighing at least 35 kg) with treatment-refractory post-transplant HCMV infection/disease (with or without genotypic resistance) with Ganciclovir, Valganciclovir, Cidofovir, or Foscarnet [58]. The recommended dosage of Maribavir for both adult and pediatric patients is 400 mg orally twice daily with or without food [59]. It is 40% bioavailable, and is highly protein-bound (98%), with free plasma concentrations of Maribavir approximately 100-fold lower than total plasma drug concentrations [60].
Marabivir resistance is mediated by UL97 mutations T409M, H411Y, and C480F. They occur in patients with recurrent HCMV infection while on therapy or having no response to therapy and confer moderate (H411Y) to moderately high (T409M) Maribavir resistance, with no cross-resistance to Ganciclovir, by mapping to a hinge region of the ATP-binding site of UL97 kinase. Particularly, C480F confers the highest degree of Maribavir resistance (224-fold) of any single mutation so far encountered in vivo, along with low-grade anciclovir cross-resistance (2.3-fold) [61][62].
In Figure 1 are reported chemical structures of approved antiviral drugs for HCMV treatment.

Figure 1. Structures of approved HCMV antivirals: (a) Ganciclovir; (b) Cidofovir; (c) Foscarnet; (d) Letermovir; (e) Maribavir.
Tomeglovir/BAY 38-4766 (C23H27N3O4S). Tomeglovir is a substituted 4-sulphonamide naphthalene derivative with good in vitro activity, which acts as a non-nucleoside inhibitor against laboratory and clinically adapted strains of HCMV through activity against the gene products UL89 and UL56. Its mechanism of action involves the prevention of viral DNA maturation during the replicative process by inhibition of viral DNA cleavage and capsid packaging [63][64]. It is under investigation, and preliminary studies showed antiviral effects in murine models and guinea pigs comparable to Ganciclovir [64][65]. Studies on the safety and tolerability of single oral doses (up to 2000 mg) of Tomeglovir were conducted in healthy male volunteers with no significant adverse events observed. Strains of drug-resistant HCMV generated by in vitro passage in the presence of Tomeglovir showed mutations in the UL89 and UL104 genes, suggesting that this new class of non-nucleoside compounds inhibits HCMV by preventing the cleavage of polygenic concatameric viral DNA into unit length genomes [66].
2-bromo-5,6-dichloro-1-(β-d-ribofuranosyl)benzimidazole (BDCRB, C7H3BrCl2N2). BDCRB and its 2-chloro homolog, 2,5,6-trichloro-1-β-d-ribofuranosyl-1H-benzimidazole (TCRB), are nucleoside analogs active against HCMV [67]. Unlike most of the currently marketed anti-HCMV agents, BDCRB and TCRB do not inhibit viral DNA synthesis, even at concentrations that completely prevent the generation of infectious virus, but exert antiviral activity by inhibition of HCMV DNA maturation. The mechanism of action is not fully understood but involves UL89 and UL56 gene products [68][69]. A study performed on guinea pig cytomegalovirus (GPHCMV) showed that the terminal structure of genomes formed in the presence of BDCRB was altered, thereby resulting in premature cleavage events and consequently in truncated genomes packed within capsids [70]. However, clinical development was not pursued after preclinical pharmacokinetic studies demonstrated that both BDCRB and TCRB are cleaved in vivo to produce the less active but more cytotoxic aglycones [68]. Within the class of benzimidazole ribosides, a derivate of BDCRB, GW275175X, exhibits similar antiviral activity without in vivo stability concerns. It acts by blocking the maturational cleavage of high-molecular-weight HCMV DNA by interaction with pUL56 and pUL89 and was advanced to Phase I clinical trial with an increasing dose of safety, pharmacokinetics, and tolerability but was later shelved to prioritize testing with Maribavir. The clinical potential of this antiviral drug still requires further study [71].
In Figure 2 are reported chemical structures of proposed terminase inhibitors for HCMV treatment.
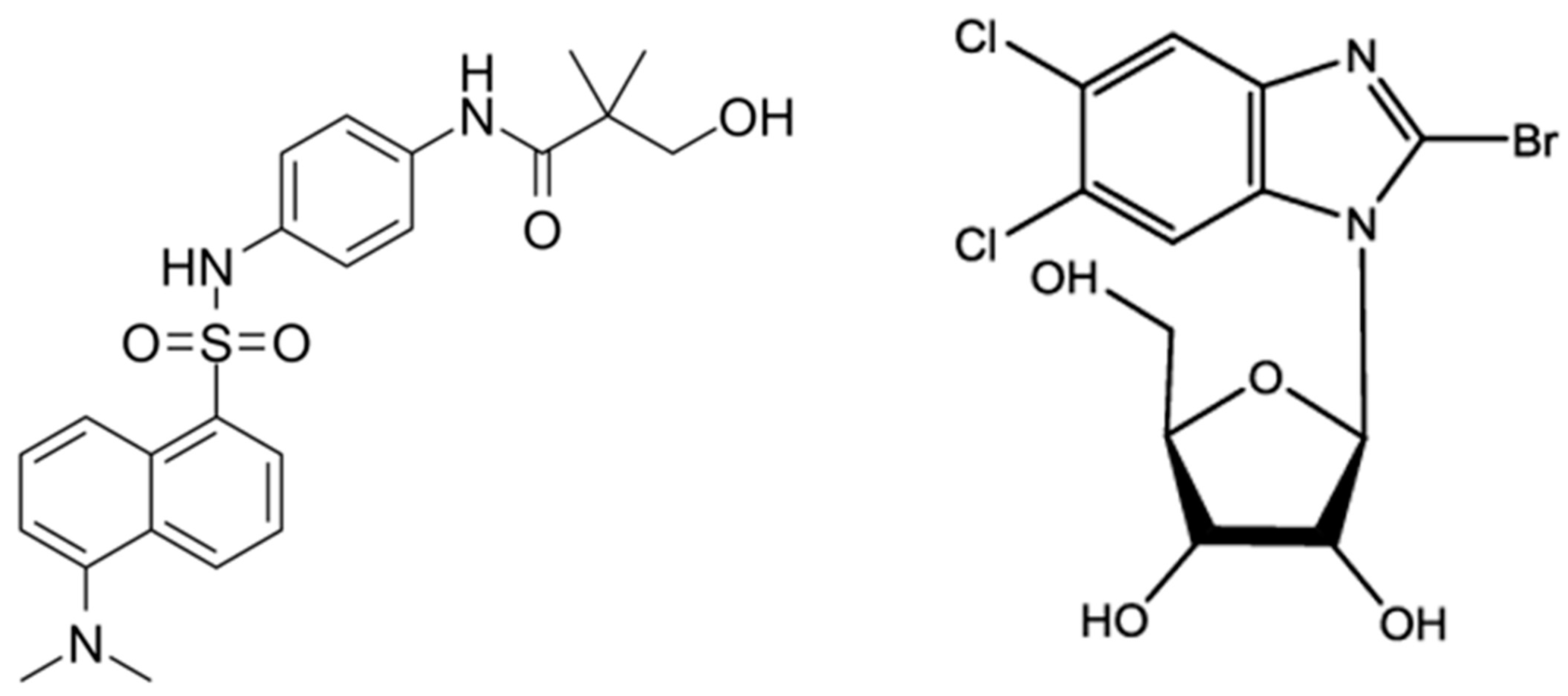
Figure 2. Structures of potential inhibitors of HCMV terminase complex: Tomeglovir (left); BDCRD (right).
In Table 1 are clinical details related to conventional and novel antiviral drugs.
Table 1. Clinical features of antiviral drugs for HCMV infection.
| Drug | Class | Status | Commercial Name | Mechanism of Action | Route of Administration |
Posology | Resistance Mechanism |
Side Effects |
|---|---|---|---|---|---|---|---|---|
| Ganciclovir | Purine nucleoside |
Clinical use (first line), FDA approval (1989) |
Cytovene® | Competitive inhibition of viral DNA polymerase | Intravenous | Induction: 1.25 mg/Kg to 5.0 mg/Kg, twice daily (7 to 14 days) Maintenance: 0.625 to 5.0 mg/Kg die Prophylaxis: 5.0 mg/Kg die (7 days per week) of 6.0 mg/kg die (5 days per week) |
Mutations on UL97 kinase and UL54 DNA polymerase genes |
Bone marrow suppression (leukopenia) |
| Valganciclovir | Purine nucleoside, modified to improve oral bioavailability |
Clinical use (first line), FDA approval (2001) |
Valcyte® | Competitive inhibition of viral DNA polymerase | Oral | Induction: 900 mg twice daily (21 days) Maintenance: 900 mg once daily Prophylaxis: 900 mg/Kg once daily |
Mutations on UL97 kinase and UL54 DNA polymerase genes |
Bone marrow suppression (leukopenia) |
| Cidofovir | Purine nucleoside |
Clinical use for treatment of HCMV ganciclovir-resistant, FDA approval (1996) |
Vistide® | Competitive inhibition of viral DNA polymerase | Intravenous, in combination with oral probenecid |
Induction: 5.0 mg/Kg, week (per 14 days) Maintenance: 5.0 mg/Kg, week Prophylaxis: not recommended |
Mutations on UL54 DNA polymerase, added to pre-existing UL97kinase mutations | Nephrotoxicity |
| Brincidofovir | Purine nucleoside |
Phase III trials, discontinued | NA | Competitive inhibition of viral DNA polymerase | Oral | NA | NA | Gastrointestinal, elevations of serum transaminases |
| Foscarnet | Pyrophosphate analog | Clinical use for treatment of HCMV ganciclovir-resistant, FDA approval (1991) |
Foscavir® | Noncompetitive inhibition of viral DNA polymerase (All Herpesvirus) |
Intravenous | Induction: 60 mg/Kg every 8 h or 90 mg/Kg, every 12 h (14 to 21 days) Maintenance: 90–120 mg/Kg die Prophylaxis: not recommended |
Mutations on UL54 DNA polymerase, cross-resistance with Ganciclovir, Valganciclovir and Cidofovir |
Nephrotoxicity, myelosuppression, mucosal ulcerations, electrolyte alterations |
| Letermovir | Quinazoline derivative |
Clinical use, FDA approval (2017) |
Prevymis® | Inhibition of viral terminase enzyme complex | Oral, intravenous | Induction: not Recommended Maintenance: not Recommended Prophylaxis: 480 mg once daily (0–28 to 100 days after transplantation) |
Mutations on UL56, UL51 and UL89 genes | Gastrointestinal |
| Maribavir | Benzimidazole riboside | Clinical use for treatment of HCMV Ganciclovir, Vfoscarnetalganciclovir, Cidofovir or Foscarnet resistant, FDA approval (2021) |
Livtencity® | Competitive inhibition of viral kinase | Oral | Post-transplant HCMV infection/disease refractory to treatment (with or without genotypic resistance): 400 mg twice daily | Mutations on UL97 kinase gene |
Gastrointestinal, dysgeusia |
| Tomeglovir | Naphthalene derivative |
Phase II trials, discontinued | NA | Inhibition of viral terminase enzyme complex | Oral | NA | NA | NA |
| BDCRD * | Benzimidazole riboside |
Phase I trials, discontinued | NA | Inhibition of viral terminase enzyme complex | NA | NA | NA | NA |
* 2-bromo-5,6-dichloro-1-(β-d-ribofuranosyl)benzimidazole; NA: not available.
This entry is adapted from the peer-reviewed paper 10.3390/microorganisms11102372
References
- Griffiths, P.; Reeves, M. Pathogenesis of human cytomegalovirus in the immunocompromised host. Nat. Rev. Microbiol. 2021, 19, 759–773.
- Ross, S.A.; Novak, Z.; Pati, S.; Boppana, S.B. Diagnosis of Cytomegalovirus Infections. Infect. Disord. Drug Targets 2011, 11, 466.
- Van Damme, E.; Van Loock, M. Functional annotation of human cytomegalovirus gene products: An update. Front. Microbiol. 2014, 5, 218.
- Ye, L.; Qian, Y.; Yu, W.; Guo, G.; Wang, H.; Xue, X. Functional Profile of Human Cytomegalovirus Genes and Their Associated Diseases: A Review. Front. Microbiol. 2020, 11, 2104.
- Bhella, D.; Rixon, F.J.; Dargan, D.J. Cryomicroscopy of human cytomegalovirus virions reveals more densely packed genomic DNA than in herpes simplex virus type 1. J. Mol. Biol. 2000, 295, 155–161.
- Yu, X.; Jih, J.; Jiang, J.; Hong Zhou, Z. Atomic structure of the human cytomegalovirus capsid with its securing tegument layer of pp150 HHS Public Access. Science 2017, 356, eaam6892.
- Butcher, S.J.; Aitken, J.; Mitchell, J.; Gowen, B.; Dargan, D.J. Structure of the human cytomegalovirus B capsid by electron cryomicroscopy and image reconstruction. J. Struct. Biol. 1998, 124, 70–76.
- Tomtishen, J. Human cytomegalovirus tegument proteins (pp65, pp71, pp150, pp28). Virol. J. 2012, 9, 22.
- Kalejta, R.F. Tegument Proteins of Human Cytomegalovirus. Microbiol. Mol. Biol. Rev. 2008, 72, 249–265.
- Garoff, H.; Hewson, R.; Opstelten, D.-J.E. Virus Maturation by Budding. Microbiol. Mol. Biol. Rev. 1998, 62, 1171.
- Homman-Loudiyi, M.; Hultenby, K.; Britt, W.; Söderberg-Nauclér, C. Envelopment of Human Cytomegalovirus Occurs by Budding into Golgi-Derived Vacuole Compartments Positive for gB, Rab 3, Trans-Golgi Network 46, and Mannosidase II. J. Virol. 2003, 77, 3191–3203.
- Foglierini, M.; Marcandalli, J.; Perez, L. HCMV envelope glycoprotein diversity demystified. Front. Microbiol. 2019, 10, 1005.
- Jean Beltran, P.M.; Cristea, I.M. The life cycle and pathogenesis of human cytomegalovirus infection: Lessons from proteomics. Expert Rev. Proteom. 2014, 11, 697–711.
- Gerna, G.; Kabanova, A.; Lilleri, D. Human cytomegalovirus cell tropism and host cell receptors. Vaccines 2019, 7, 70.
- Isomura, H.; Stinski, M.F.; Murata, T.; Yamashita, Y.; Kanda, T.; Toyokuni, S.; Tsurumi, T. The Human Cytomegalovirus Gene Products Essential for Late Viral Gene Expression Assemble into Prereplication Complexes before Viral DNA Replication. J. Virol. 2011, 85, 6629–6644.
- Garci´a, J.J.; Garci´a-Rami´rez, G.; Rami´rez, R.; Ruchti, F.; Huang, H.; Simmen, K.; Angulo, A.; Ghazal, P. Dominance of Virus over Host Factors in Cross-Species Activation of Human Cytomegalovirus Early Gene Expression. J. Virol. 2001, 75, 26–35.
- Adamson, C.S.; Nevels, M.M. Bright and early: Inhibiting human cytomegalovirus by targeting major immediate-early gene expression or protein function. Viruses 2020, 12, 110.
- Omoto, S.; Mocarski, E.S. Transcription of True Late (γ2) Cytomegalovirus Genes Requires UL92 Function That Is Conserved among Beta- and Gammaherpesviruses. J. Virol. 2014, 88, 120–130.
- Rozman, B.; Nachshon, A.; Levi Samia, R.; Lavi, M.; Schwartz, M.; Stern-Ginossar, N. Temporal dynamics of HCMV gene expression in lytic and latent infections. Cell Rep. 2022, 39, 110653.
- Crough, T.; Khanna, R. Immunobiology of human cytomegalovirus: From bench to bedside. Clin. Microbiol. Rev. 2009, 22, 76–98.
- Poole, E.; Wills, M.; Sinclair, J. Human Cytomegalovirus Latency: Targeting Differences in the Latently Infected Cell with a View to Clearing Latent Infection. New J. Sci. 2014, 2014, 1–10.
- Cannon, M.J.; Schmid, D.S.; Hyde, T.B. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol. 2010, 20, 202–213.
- Fowler, K.; Mucha, J.; Neumann, M.; Lewandowski, W.; Kaczanowska, M.; Grys, M.; Schmidt, E.; Natenshon, A.; Talarico, C.; Buck, P.O.; et al. A systematic literature review of the global seroprevalence of cytomegalovirus: Possible implications for treatment, screening, and vaccine development. BMC Public Health 2022, 22, 1659.
- Chen, S.J.; Wang, S.C.; Chen, Y.C. Antiviral agents as therapeutic strategies against cytomegalovirus infections. Viruses 2019, 12, 21.
- Panda, K.; Parashar, D.; Viswanathan, R. An Update on Current Antiviral Strategies to Combat Human Cytomegalovirus Infection. Viruses 2023, 15, 1358.
- Navarro, D.; San-Juan, R.; Manuel, O.; Giménez, E.; Fernández-Ruiz, M.; Hirsch, H.H.; Grossi, P.A.; Aguado, J.M. Cytomegalovirus infection management in solid organ transplant recipients across European centers in the time of molecular diagnostics: An ESGICH survey. Transpl. Infect. Dis. 2017, 19, e12773.
- Yadav, D.K.; Adhikari, V.P.; Yadav, R.K.; Singh, A.; Huang, X.; Zhang, Q.; Pandit, P.; Ling, Q.; Liang, T. Antiviral prophylaxis or preemptive therapy for cytomegalovirus after liver transplantation?: A systematic review and meta-analysis. Front. Immunol. 2022, 13, 953210.
- Razonable, R.R.; Humar, A. Cytomegalovirus in solid organ transplant recipients—Guidelines of the American Society of Transplantation Infectious Diseases Community of Practice. Clin. Transplant. 2019, 33, e13512.
- Grossi, P.A.; Baldanti, F.; Andreoni, M.; Perno, C.F. CMV infection management in transplant patients in Italy. J. Clin. Virol. 2020, 123, 104211.
- Markham, A.; Faulds, D. Ganciclovir. An update of its therapeutic use in cytomegalovirus infection. Drugs 1994, 48, 455–484.
- Crumpacker, C.S. Ganciclovir. N. Engl. J. Med. 1996, 335, 721–729.
- Galar, A.; Valerio, M.; Catalán, P.; García-González, X.; Burillo, A.; Fernández-Cruz, A.; Zataráin, E.; Sousa-Casasnovas, I.; Anaya, F.; Rodríguez-Ferrero, M.L.; et al. Valganciclovir—Ganciclovir use and systematic therapeutic drug monitoring. An invitation to antiviral stewardship. Antibiotics 2021, 10, 77.
- Kotton, C.N.; Kumar, D.; Caliendo, A.M.; Åsberg, A.; Chou, S.; Snydman, D.R.; Allen, U.; Humar, A.; Emery, V.; Lautenschlager, I.; et al. International consensus guidelines on the management of cytomegalovirus in solid organ transplantation. Transplantation 2010, 89, 779–795.
- Girmenia, C.; Lazzarotto, T.; Bonifazi, F.; Patriarca, F.; Irrera, G.; Ciceri, F.; Aversa, F.; Citterio, F.; Cillo, U.; Cozzi, E.; et al. Assessment and prevention of cytomegalovirus infection in allogeneic hematopoietic stem cell transplant and in solid organ transplant: A multidisciplinary consensus conference by the Italian GITMO, SITO, and AMCLI societies. Clin. Transplant. 2019, 33, e13666.
- Samuel, E.; McNaught, K.A.; Mulbah, J.L.; HajiAlilou, H.; Mody, V.; Cates, D.W. Antiviral drugs. In Side Effects of Drugs Annual; Elsevier: Amsterdam, The Netherlands, 2022; Volume 44, pp. 291–301. ISBN 9780323989091.
- Boonsathorn, S.; Pasomsub, E.; Techasaensiri, C.; Apiwattanakul, N. Analysis of Ganciclovir-Resistant Cytomegalovirus Infection Caused by the UL97 Gene Mutation in Codons 460 and 520 in Pediatric Patients: A Case Series. Open Forum Infect. Dis. 2019, 6, ofz480.
- Das, D.; Hong, J. Herpesvirus Polymerase Inhibitors. In Viral Polymerases; Academic Press: Cambridge, MA, USA, 2019; pp. 333–356.
- Lea, A.P.; Bryson, H.M. Cidofovir. Drugs 1996, 52, 225–230.
- Paintsil, E.; Cheng, Y.C. Antiviral Agents. In Encyclopedia of Microbiology; Academic Press: Cambridge, MA, USA, 2009; pp. 223–257.
- Meesing, A.; Razonable, R.R. New Developments in the Management of Cytomegalovirus Infection after Transplantation. Drugs 2018, 78, 1085–1103.
- Chou, S.; Komazin-Meredith, G.; Williams, J.D.; Bowlin, T.L. Cytomegalovirus mutants resistant to ganciclovir and cidofovir differ in susceptibilities to synguanol and its 6-ether and 6-thioether derivatives. Antimicrob. Agents Chemother. 2014, 58, 1809–1812.
- Bogner, E.; Egorova, A.; Makarov, V. Small Molecules—Prospective Novel HCMV Inhibitors. Viruses 2021, 13, 474.
- Salvaggio, M.R.; Gnann, J.W. Drugs for Herpesvirus Infections. In Infectious Diseases; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1309–1317.e1.
- Wagstaff, A.J.; Bryson, H.M. Foscarnet: A reappraisal of its antiviral activity, pharmacokinetic properties and therapeutic use in immunocompromised patients with viral infections. Drugs 1994, 48, 199–226.
- Avery, R.K.; Arav-Boger, R.; Marr, K.A.; Kraus, E.; Shoham, S.; Lees, L.; Trollinger, B.; Shah, P.; Ambinder, R.; Neofytos, D.; et al. Outcomes in Transplant Recipients Treated with Foscarnet for Ganciclovir-Resistant or Refractory Cytomegalovirus Infection. Transplantation 2016, 100, e74–e80.
- Chrisp, P.; Clissold, S.P. Foscarnet: A review of its antiviral activity, pharmacokinetic properties and therapeutic use in immunocompromised patients with cytomegalovirus retinitis. Drugs 1991, 41, 104–129.
- Chou, S. Foscarnet resistance mutations mapping to atypical domains of the cytomegalovirus DNA polymerase gene. Antivir. Res. 2017, 138, 57.
- Kimberlin, D.W. Antiviral Agents. In Principles and Practice of Pediatric Infectious Diseases; Elsevier: Amsterdam, The Netherlands, 2023; pp. 1583–1598.e6.
- Lischka, P.; Hewlett, G.; Wunberg, T.; Baumeister, J.; Paulsen, D.; Goldner, T.; Ruebsamen-Schaeff, H.; Zimmermann, H. In vitro and in vivo activities of the novel anticytomegalovirus compound AIC246. Antimicrob. Agents Chemother. 2010, 54, 1290–1297.
- Melendez, D.P.; Razonable, R.R. Letermovir and inhibitors of the terminase complex: A promising new class of investigational antiviral drugs against human cytomegalovirus. Infect. Drug Resist. 2015, 8, 269.
- Raglow, Z.; Kaul, D.R. A New Antiviral Option for Cytomegalovirus Prevention after Kidney Transplant. JAMA 2023, 330, 27–29.
- Shigle, T.L.; Handy, V.W.; Chemaly, R.F. Letermovir and its role in the prevention of cytomegalovirus infection in seropositive patients receiving an allogeneic hematopoietic cell transplant. Ther. Adv. Hematol. 2020, 11, 204062072093715.
- El Helou, G.; Razonable, R.R. Letermovir for the prevention of cytomegalovirus infection and disease in transplant recipients: An evidence-based review. Infect. Drug Resist. 2019, 12, 1481–1491.
- Santos Bravo, M.; Tilloy, V.; Plault, N.; Palomino, S.S.; Mosquera, M.M.; Navarro Gabriel, M.; Fernández Avilés, F.; Suárez Lledó, M.; Rovira, M.; Moreno, A.; et al. Assessment of UL56 Mutations before Letermovir Therapy in Refractory Cytomegalovirus Transplant Recipients. Microbiol. Spectr. 2022, 10, e00191-22.
- Hofmann, E.; Sidler, D.; Dahdal, S.; Bittel, P.; Suter-Riniker, F.; Manuel, O.; Walti, L.N.; Hirzel, C. Emergence of letermovir resistance in solid organ transplant recipients with ganciclovir resistant cytomegalovirus infection: A case series and review of the literature. Transpl. Infect. Dis. 2021, 23, e13515.
- Halpern-Cohen, V.; Blumberg, E.A. New Perspectives on Antimicrobial Agents: Maribavir. Antimicrob. Agents Chemother. 2022, 66, e02405-21.
- Avery, R.K.; Alain, S.; Alexander, B.D.; Blumberg, E.A.; Chemaly, R.F.; Cordonnier, C.; Duarte, R.F.; Florescu, D.F.; Kamar, N.; Kumar, D.; et al. Maribavir for Refractory Cytomegalovirus Infections with or without Resistance Post-Transplant: Results From a Phase 3 Randomized Clinical Trial. Clin. Infect. Dis. 2022, 75, 690–701.
- Kang, C. Maribavir: First Approval. Drugs 2022, 82, 335–340.
- Maertens, J.; Cordonnier, C.; Jaksch, P.; Poiré, X.; Uknis, M.; Wu, J.; Wijatyk, A.; Saliba, F.; Witzke, O.; Villano, S. Maribavir for Preemptive Treatment of Cytomegalovirus Reactivation. N. Engl. J. Med. 2019, 381, 1136–1147.
- Wang, L.H.; Peck, R.W.; Yin, Y.; Allanson, J.; Wiggs, R.; Wire, M.B. Phase I safety and pharmacokinetic trials of 1263W94, a novel oral anti-human cytomegalovirus agent, in healthy and human immunodeficiency virus-infected subjects. Antimicrob. Agents Chemother. 2003, 47, 1334–1342.
- Chou, S.; Alain, S.; Cervera, C.; Chemaly, R.F.; Kotton, C.N.; Lundgren, J.; Papanicolaou, G.A.; Pereira, M.R.; Wu, J.J.; Murray, R.A.; et al. Drug Resistance Assessed in a Phase 3 Clinical Trial of Maribavir Therapy for Refractory or Resistant Cytomegalovirus Infection in Transplant Recipients. J. Infect. Dis. 2023, jiad293.
- Chou, S.; Song, K.; Wu, J.; Bo, T.; Crumpacker, C. Drug Resistance Mutations and Associated Phenotypes Detected in Clinical Trials of Maribavir for Treatment of Cytomegalovirus Infection. J. Infect. Dis. 2022, 226, 576–584.
- Komazin, G.; Townsend, L.B.; Drach, J.C. Role of a Mutation in Human Cytomegalovirus Gene UL104 in Resistance to Benzimidazole Ribonucleosides. J. Virol. 2004, 78, 710–715.
- Schleiss, M.R.; Bernstein, D.I.; Mcvoy, M.A.; Stroup, G.; Bravo, F.; Creasy, B.; Mcgregor, A.; Henninger, K.; Hallenberger, S. The non-nucleoside antiviral, BAY 38-4766, protects against cytomegalovirus (CMV) disease and mortality in immunocompromised guinea pigs. Antivir. Res. 2005, 65, 35–43.
- Weber, O.; Bender, W.; Eckenberg, P.; Goldmann, S.; Haerter, M.; Hallenberger, S.; Henninger, K.; Reefschläger, J.; Trappe, J.; Witt-Laido, A.; et al. Inhibition of murine cytomegalovirus and human cytomegalovirus by a novel non-nucleosidic compound in vivo. Antivir. Res. 2001, 49, 179–189.
- Kosobucki, B.R.; Freeman, W.R. Retinal Disease in HIV-infected Patients. In Retina, 4th ed.; Mosby: Maryland Heights, MO, USA, 2006; Volume 2–3, pp. 1625–1672.
- Townsend, L.B.; Devivar, R.V.; Turk, S.R.; Nassiri, M.R.; Drach, J.C. Design, synthesis, and antiviral activity of certain 2,5,6-trihalo-1-(beta-D-ribofuranosyl)benzimidazoles. J. Med. Chem. 1995, 38, 4098–4105.
- Underwood, M.R.; Ferris, R.G.; Selleseth, D.W.; Davis, M.G.; Drach, J.C.; Townsend, L.B.; Biron, K.K.; Boyd, F.L. Mechanism of action of the ribopyranoside benzimidazole GW275175X against human cytomegalovirus. Antimicrob. Agents Chemother. 2004, 48, 1647–1651.
- McVoy, M.A.; Nixon, D.E. Impact of 2-Bromo-5,6-Dichloro-1-β-d-Ribofuranosyl Benzimidazole Riboside and Inhibitors of DNA, RNA, and Protein Synthesis on Human Cytomegalovirus Genome Maturation. J. Virol. 2005, 79, 11115–11127.
- Nixon, D.E.; McVoy, M.A. Dramatic effects of 2-bromo-5,6-dichloro-1-beta-D-ribofuranosyl benzimidazole riboside on the genome structure, packaging, and egress of guinea pig cytomegalovirus. J. Virol. 2004, 78, 1623–1635.
- Gugliesi, F.; Coscia, A.; Griffante, G.; Galitska, G.; Pasquero, S.; Albano, C.; Biolatti, M. Where do we stand after decades of studying human cytomegalovirus? Microorganisms 2020, 8, 685.
This entry is offline, you can click here to edit this entry!