Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Microbiology
Crimean–Congo hemorrhagic fever virus (CCHFV), a member of the Nairoviridae family and Bunyavirales order, is transmitted to humans via tick bites or contact with the blood of infected animals. It can cause severe symptoms, including hemorrhagic fever, with a mortality rate between 5 to 30%. CCHFV is classified as a high-priority pathogen by the World Health Organization (WHO) due to its high fatality rate and the absence of effective medical countermeasures.
- Crimean–Congo hemorrhagic fever virus
- hemorrhagic fever
- immune response
1. Introduction
Crimean–Congo hemorrhagic fever virus (CCHFV) is a member of the genus Orthonairovirus in the family Nairoviridae and the order Bunyavirales [1,2]. The viral genome is composed of three negative-sense RNA segments: small (S), medium (M), and large (L). The S segment encodes the nucleoprotein (NP), while the M segment encodes the glycoprotein precursor (GPC), which later forms mature Gn, Gc, and several nonstructural proteins such as mucin, GP38, and NSm [3,4,5,6]. The L segment encodes the L protein responsible for viral RNA synthesis, which includes the RNA-dependent RNA polymerase (RdRp) and an ovarian tumor (OTU) protease domain (for which the L protein is crucial for viral RNA synthesis) and includes the RNA-dependent RNA polymerase (RdRp) and an OTU protease domain that may aid in evading the host’s innate immunity (Figure 1a,b) [7,8,9].
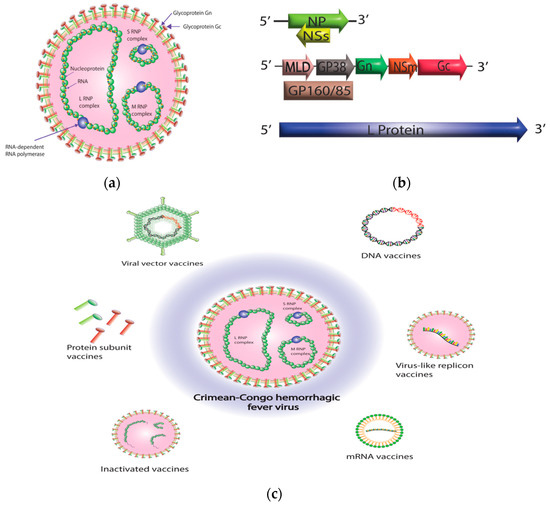
Figure 1. Crimean–Congo hemorrhagic fever virus (CCHFV) virion structure and CCHFV vaccine platforms. (a) CCHFV virion contains three single-stranded RNA segments with a negative-sense orientation. Nucleoprotein (NP) and RNA-dependent RNA polymerase (RdRp; L protein) protect the RNA by enclosing the RNA segments and forming ribonucleoprotein complexes (RNPs). Once the ribonucleoprotein (RNP) complexes are formed, they are surrounded by a protective envelope that originates from the membrane of the host cell. This envelope is coated with specialized glycoproteins known as Gn and Gc. (b) CCHFV consists of three genomic segments—small (S), medium (M), and large (L). The S segment is responsible for encoding the NP within one open reading frame, while the small non-structural protein (NSs) is encoded in an opposite-sense open reading frame. The M segment is quite intricate, as it encodes a glycoprotein precursor (GPC) that undergoes processing by host proteases. This processing results in the production of a GP160/85 domain, which is then further processed into a mucin-like domain (MLD) and GP38. Additionally, the M segment encodes the Gn and Gc glycoproteins, as well as the medium non-structural protein (NSm). The L segment of CCHFV, which is distinctively larger than other bunyaviruses, encodes for the viral RNA-dependent RNA polymerase (RdRP) and an ovarian tumor-like protease (OTU) at its N terminus. (c) A diagrammatic representation of different CCHFV vaccine platforms. The diagram was created with Adobe Illustrator.
It was initially detected in Soviet soldiers in Crimea during the 1940s. In the 1960s, a virus with similar symptoms to the Crimean virus was discovered in the Belgian Congo (currently known as the Democratic Republic of the Congo). Further studies revealed that both viruses were antigenically identical, leading to the virus being named CCHFV [10,11,12,13]. CCHFV circulates within an enzootic cycle that involves ticks and vertebrates [14,15,16]. Although CCHFV has been isolated in multiple tick species, Hyolamma ticks serve as the host and biological vector for CCHFV due to its extensive geographical range, which closely correlates with the distribution of CCHF cases [17,18]. Ticks can transmit CCHFV vertically from one generation to the next, transovarially from one developmental stage to another, sexually from males to females during copulation, or through cofeeding from one tick to other ticks feeding on the same non-viremic host [19,20,21,22]. CCHFV infects a wide range of both wild and domestic animals. However, infections in these animals are typically asymptomatic, but they exhibit viremia for more than five days, which helps the maintenance of CCHFV in nature [14,17,18,19,20,21,22].
CCHFV is found in a vast geographic region, from western China to Africa, the Middle East, Spain, and the Balkans [23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40]. Turkey has been experiencing CCHF epidemics since 2002, with the number of cases increasing significantly in recent years [41,42,43,44,45]. Similarly, some Balkan countries have reported regular cases of the disease [46]. CCHF was first reported in Spain in 2016, although tick surveillance studies had already shown the presence of CCHFV in the area [36]. This suggests a shift in the main vector’s geographic distribution, as there were no previous reports of autochthonous human cases in Spain [47,48,49]. Ticks primarily disperse over long distances only through their hosts [50]. Therefore, changes in tick populations are primarily associated with bird migrations or expansions of host populations. The geographical spread of tick populations is concerning as infected ticks transported to non-endemic areas can spread the disease to humans [48,51,52,53]. Furthermore, uninfected ticks introduced to a new area may establish populations that can sustain the virus after its introduction. The expansion of CCHF’s geographic distribution is also driven by several factors, including global warming, the increasing of human mobility, and human activities such as deforestation and agricultural growth, leading to more people coming into contact with infected ticks and animals [54,55,56,57]. Transporting livestock is a widespread practice across the world, and its contribution to the spread of CCHF cannot be overlooked. The movement of animals across borders or the transportation of infected ticks from endemic regions can initiate new CCHF outbreaks in non-endemic regions [58,59,60,61,62].
Human beings are regarded as accidental hosts of the CCHFV. Humans become infected through tick bites or exposure to crushed infected ticks during agricultural activities. Another significant source of infection is the blood of infected agricultural animals, which can be viremic but not display any symptoms of disease [14,17]. Nosocomial transmission contributes to the spread of CCHFV, leading to higher fatality rates compared to those resulting from tick bites. Several cases of nosocomial outbreaks have been linked to infected blood or needle-stick injuries during patient care [63,64,65,66,67]. CCHFV infection in humans can cause mild to severe symptoms, including high fever, malaise, myalgia, and gastrointestinal distress, typically after a short incubation period of about a week. Severe cases can result in hemorrhagic disease with a fatality rate ranging from 5 to 30%, often due to disseminated intravascular coagulopathy, shock, and/or multi-organ failure [63,64,65,67]. Due to its high fatality rate, widespread vector, and the absence of effective medical countermeasures for prevention and treatment, CCHFV is classified as a high-priority pathogen by the World Health Organization (WHO) [68].
2. Immune Response to CCHF Infection
Effective control of CCHFV infection in the host relies on immune responses from both the innate and adaptive systems. The innate immune system acts as the first line of defense against viruses by limiting viral entry, translation, replication, and assembly. Additionally, it facilitates the identification and elimination of infected cells, as well as the development of adaptive immunity through coordination and acceleration. Type 1 interferons (IFN-α/β) are produced by the host’s innate immune response against viruses. These responses are rapid and efficient, and they can be generated and secreted by all mammalian cells. These immunomodulators facilitate the expression of antiviral proteins, inhibit cell proliferation, and help regulate apoptosis [69,70,71].
The innate immune response is stimulated by CCHFV, leading to the production of IFNs and interferon-stimulated genes (ISGs). Andersson et al. conducted in vitro studies that confirmed the antiviral effect of IFN on CCHFV. The studies highlighted a substantial reduction of vRNA levels in cells treated with IFN, emphasizing the crucial role of IFN in controlling CCHFV replication [72]. Hawman et al. recently created a novel model using type I interferon-deficient mice, whereby infection with the human clinical isolate strain Hoti resulted in progressive illness characterized by several days of overt clinical signs [73]. This model also demonstrated the induction and release of IFNs, the subsequent upregulation of ISGs, and the involvement of the host’s innate immune response to CCHFV [74]. Bente et al. confirmed the crucial role of IFN in combating CCHFV using a STAT-1 KO mouse model, where STAT1 is a central component of IFN signaling pathways [75]. In 2012, CCHFV-infected IFN1-deficient mice (IFNAR−/−) exhibited clinical symptoms resembling CCHF, whereas wild-type mice remained asymptomatic, highlighting the importance of IFN1 in preventing CCHFV infection [76]. IFN is also crucial in controlling infections and preventing diseases in animals, even those with compromised adaptive immune systems.
Lindquist et al. demonstrated the temporary suppression of the immune response in various mouse strains (including wild-type and those with impaired adaptive immunity such as NOD/SCID, Prf1−/− and Rag2−/−) by utilizing an anti-IFNAR1 monoclonal antibody (mAb) [77]. IFN responses play a significant role in determining disease severity. Studies indicated that polymorphisms in toll-like receptor genes (TLR7, 8, 9, and 10) are associated with increased illness severity in Turkish CCHF patients, thereby emphasizing the crucial role of TLRs as an immune-sensing pathway in controlling the virus [78,79]. Despite IFNs being crucial in the host’s immune response, CCHFV utilizes various strategies to evade and counteract the innate immune response. These include removing the 5’ triphosphate group from the viral genome to avoid RIG-I recognition, delaying IRF-3 activation through the particle recognition pathway, and downregulating NF-kappaB activation [80,81,82]. Additionally, studies have shown that CCHFV can suppress the body’s innate immune system by encoding an ovarian tumor-related deubiquitinase (OTU) domain. This domain deubiquitinates proteins involved in the body’s signaling pathways, thereby inhibiting innate immune responses including the antiviral response mediated by ISG15 modifications. In addition, the OTU domain has both de-ISGylation and deubiquitinase activity, which are important for viral pathogenesis [8,83].
Apoptosis can serve as a significant innate response to viral infections. It is a critical process involved in viral infections, as its suppression or induction determines the level of infection spread. Viruses can either inhibit host cell apoptosis, which is a defense mechanism against infections, to ensure their survival or on the contrary promote apoptosis to eliminate uninfected immune cells and facilitate viral spread [84,85]. As with many other viruses, CCHFV also has the ability to regulate apoptosis. CCHF infection induces TNF-α and FasL-mediated apoptosis in cell culture [86]. CCHFV NP inhibits caspase 3 and caspase 9 activation, prevents apoptosis initiated by BAX, and curtails the release of cytochrome c from mitochondria [87]. However, the specific point in the intrinsic apoptosis pathway where NP disrupts activation is yet to be determined. Despite the CCHFV NP inhibiting activation in the intrinsic pathway of apoptosis, the CCHFV NSs, a cryptic ambisense product of the NP, has been found to disrupt mitochondrial membrane potential, inducing apoptosis by activating caspase 3/7 and cleaving poly ADP-ribose polymerase. Furthermore, the presence of a conserved DEVD motif in the virus’ NP that can be cleaved by host caspase 3 implies a regulatory role in the virus’s life cycle [88].
Pro-inflammatory cytokines and chemokines, which are produced by various immune cells in response to viral infections, play a crucial role in the innate immune response against viral pathogens [89]. When a virus enters the body, immune cells such as macrophages and dendritic cells recognize it through pattern recognition receptors (PRRs) on their surface. These PRRs identify pathogen-associated molecular patterns (PAMPs) that are unique to the invading virus. Upon recognition of viral PAMPs, immune cells release pro-inflammatory cytokines [89,90]. While pro-inflammatory cytokines and chemokines are critical for the innate immune response to viral infections, excessive or uncontrolled release of these molecules can lead to tissue damage and inflammatory disease [91,92]. CCHFV initially targets immune cells such as dendritic cells, macrophages, and monocytes [93]. This results in the production of pro-inflammatory cytokines, including TNF-α, IL1, IL6, IL8, IL-12, IFN-γ, MCP-1, and MIP-1b [94]. Pro-inflammatory responses in severe or fatal diseases can lead to vascular dysfunction, disseminated intravascular coagulation (DIC), organ failure, and shock [95]. Increased levels of TNF-α, IL-8, IL-9, IL-15, IP-10, and MCP-1 are associated with disease severity and negative outcomes in patients from Turkey, Albania, and Kosovo [17,94,95,96,97]. Researchers have also found that the secretion of sTREM-1 by myeloid cells enhances inflammatory responses during CCHF virus infection, though it remains to be empirically demonstrated how excessive levels of these inflammatory agents may drive pathogenic processes [98,99].
While innate immunity serves as the initial protective response against CCHFV infection, a potent adaptive immune response is also essential for effective control of the infection. Anti-CCHFV IgM and IgG antibodies are detectable within 7–9 days from symptom onset [100,101,102]. IgG antibodies typically peak during the second to third week and can remain for up to 3 years. In contrast, IgM titers decline within 3 weeks and become undetectable between 3–5 months after disease onset. The absence of serum antibodies has been linked to higher mortality rates in CCHF patients, suggesting that antibodies may provide protection against fatal CCHFV infection [97,100]. Kaya et al. assessed serial antibody responses on 31 patients with CCHF, 11 of which were fatal cases. The study revealed that all surviving patients had a positive IgG titer within 9 days of onset, whereas none of the fatal cases showed such a response at the same timepoint [103]. In a study conducted on 24 patients with 43 samples, it was observed that quantitative IgG levels and viral loads had a correlation, and none of the fetal patients developed positive IgG titers. Only one sample from nine survivors taken less than nine days after the onset of the disease showed positive IgG titers. There was no correlation between death or viral load and IgM positivity [104]. In a study involving 46 confirmed cases of acute CCHFV infection in Kosovo, it was discovered that there was no correlation between the presence of IgM antibodies and clinical classification. Furthermore, only 5 out of the 34 patients who survived the disease exhibited IgG antibodies [95].
Neutralizing antibodies (NAbs) usually appear by day 10 of the illness. They are usually found at low levels in CCHF survivors, but are undetectable in fatal cases of CCHF [97,100]. This suggests that antibodies may play a crucial role in protecting individuals from lethal CCHFV infections. As of now, the study has revealed that mAbs and neutralizing mAbs specific to CCHFV have exclusively been derived from mice [105]. Among the isolated antibodies, three Gc-specific NAbs have demonstrated the ability to neutralize multiple strains of the virus [105,106]. However, despite their cross-neutralizing activity, these NAbs have not been effective in providing protection against CCHF in experiments conducted on mouse models. These epitopes are not associated with the production of NAbs that contribute to the immune response against CCHF. Three Gc-specific NAbs cross-neutralize various strains but are ineffective in protecting mouse models of CCHF [105,106]. Studies have shown that although mAbs targeting pre-Gn and/or GP38 lack neutralizing activity, they can still provide pre-exposure protection in mice [105,106,107,108]. These findings suggest that non-NAbs may also have the ability to protect against CCHF through other mechanisms besides neutralization, as evidenced by their ability to confer protection in fatal CCHF challenges. The GP38-targeting mAbs effectiveness in providing protection depends on complement activity. This finding suggests that the antibody’s effector functions, such as complement-mediated lysis and phagocytosis, play a crucial role in protecting against CCHFV [107]. However, a recent study illustrated the efficacy of bispecific antibodies (bsAbs) by incorporating variable domains from wide NAbs to boost their antiviral efficacy. The study found one bsAb to be particularly effective as it provided therapeutic protection against CCHFV with a single dose [109]. Thus, it is currently unclear whether there is any relationship between the neutralization antibody responses and positive disease outcomes.
Several studies have found a strong relationship between a high viral load and fatality rate, with some identifying a viral load of ≥108 copies/mL as a significant predictor of fatality [63,95,104,110]. While a reduction in CCHF viral load has been connected with the presence of antibodies in clinical infections, the production of antibodies is not always linked to the clearance of the virus. During the first week of infection, viral loads generally decrease irrespective of IgG levels, indicating the critical role of cellular immunity [95,104]. A study conducted with STAT1-deficient mice showed that CD4+ and CD8+ T cells were activated early against CCHFV infection [75]. Studies on infected mice have demonstrated that T cells play a critical role in controlling CCHFV infection. They limit the virus’s spread and prevent further infection by producing antiviral cytokines upon rapid activation [73,74].
The maintenance of CCHFV-specific T cells for an extended period after infection suggests that memory T cells may offer long-term immunity against the virus, responding rapidly to future exposure and serving as a lasting defense [111]. The study on a DNA vaccine highlights the significance of a TH1 response for effective protection [112]. It was shown that CD8+ T cell responses in human survivors lasted for 13 years after acute infection. Most T cell epitopes were found on the NP, but there were two instances of reactivity to GC-derived peptides. None of the epitopes were considered immunodominant [113]. Lindquist et al. found that an IFN blockade antibody treatment in mice effectively controlled CCHFV through adaptive immune responses, specifically cytolytic T cell activity, while avoiding liver damage, which is a common issue caused directly by CCHFV [77]. The removal of CD4+ or CD8+ T cells in mice infected with the virus resulted in a higher mortality rate, highlighting the indispensability of these cells in survival [74]. Hawman et al. also found that the absence of CD4 T cells eliminated the host’s IFN-γ response and blocking IFN-γ signaling led to lethality in IFNAR−/− mice, suggesting that cellular immunity and type II IFN may control the CCHFV infection. Despite the existing research on the role of the adaptive immune response in CCHFV infection, further studies are needed to determine the immune responses and their effector functions essential for protection.
This entry is adapted from the peer-reviewed paper 10.3390/diagnostics13162708
This entry is offline, you can click here to edit this entry!