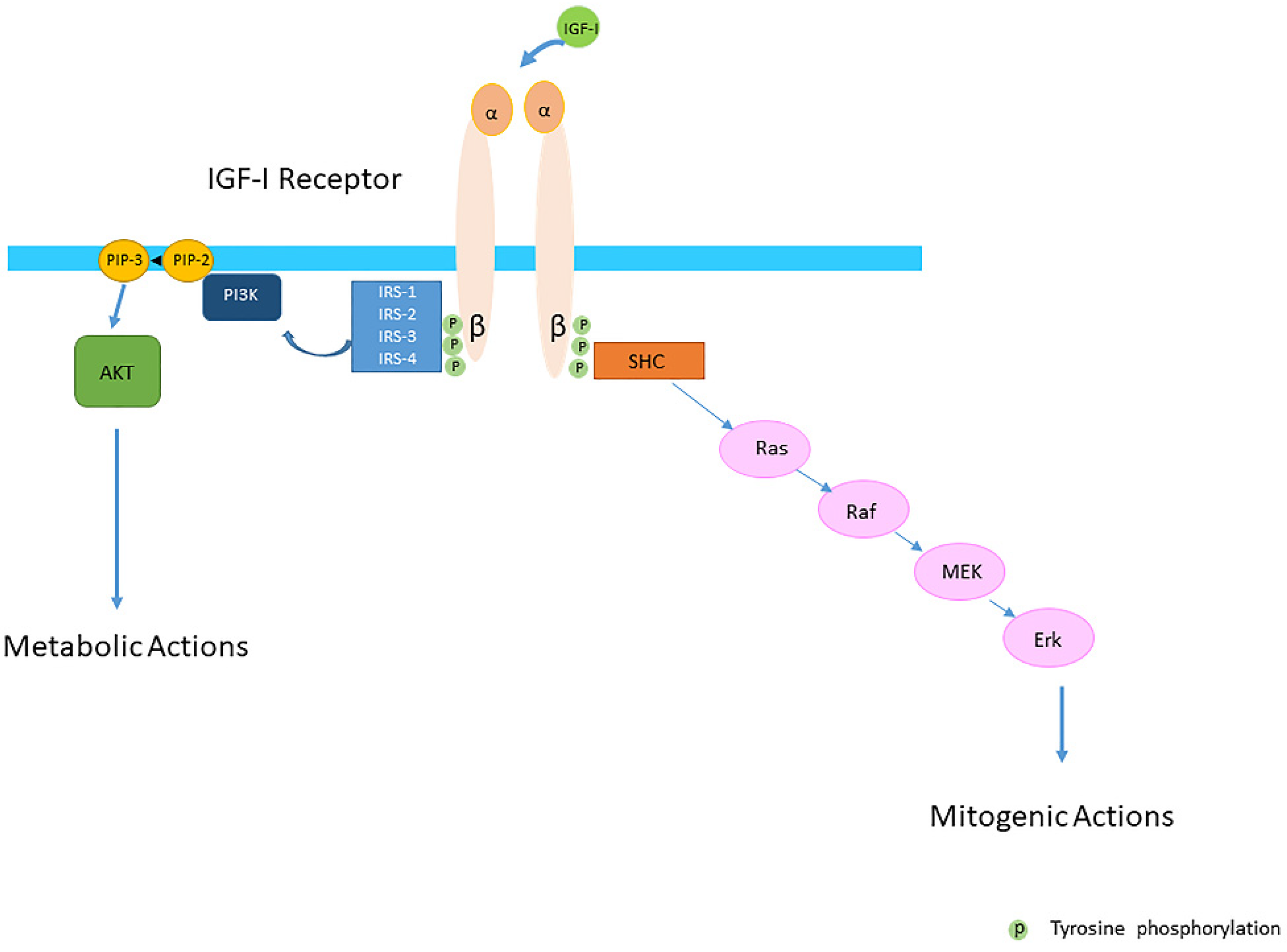
Insulin-like growth factor-I (IGF-I) and insulin-like growth factor-II (IGF-II) play a crucial factor in the growth, differentiation and survival of cells in health and disease. IGF-I and IGF-II primarily activate the IGF-I receptor (IGF-IR), which is present on the cell surface. Activation of the IGF-IR stimulates multiple pathways which finally results in multiple biological effects in a variety of tissues and cells. In addition, activation of the IGF-IR has been found to be essential for the growth of cancers. The conventional view in the past was that the IGF-IR was exclusively a tyrosine kinase receptor and that phosphorylation of tyrosine residues, after binding of IGF-I to the IGF-IR, started a cascade of post-receptor events. Research has shown that this view was too simplistic. It has been found that the IGF-IR also has kinase-independent functions and may even emit signals in the unoccupied state through some yet-to-be-defined non-canonical pathways. The IGF-IR may further form hybrids with the insulin receptors but also with receptor tyrosine kinases (RTKs) outside the insulin-IGF system. In addition, the IGF-IR has extensive cross-talk with many other receptor tyrosine kinases and their downstream effectors. Moreover, there is now emerging evidence that the IGF-IR utilizes parts of the G-protein coupled receptor (GPCR) pathways: the IGF-IR can be considered as a functional RTK/GPCR hybrid, which integrates the kinase signaling with some IGF-IR mediated canonical GPCR characteristics. Like the classical GPCRs the IGF-IR can also show homologous and heterologous desensitization.
- IGF-I
- IGF-II
- insulin
- IGF-IR
- IRs
- tyrosine kinase receptor
- GPCRs
- hybrids
- phosphorylation
- G-proteins
1. Introduction
2. IGF-I and the IGF-I Receptor
3. The IGF-IR and Endocytosis

4. Structural Differences and Overlap between the IGF-IR and the IRs
5. The IGF-IR and the IRs May Form Hybrids in the Human Body

This entry is adapted from the peer-reviewed paper 10.3390/cells9040862
References
- Collett-Solberg, P.F.; Cohen, P. The role of the insulin-like growth factor binding proteins and the igfbp proteases in modulating igf action. Endocrinol. Metab. Clin. N. Am. 1996, 25, 591–614.
- Janssen, J.; Varewijck, A.J.; Brugts, M.P. The insulin-like growth factor-i receptor stimulating activity (irsa) in health and disease. Growth Horm. Igf Res. 2019, 48–49, 16–28.
- Bach, L.A. Igf-binding proteins. J. Mol. Endocrinol. 2018, 61, T11–T28.
- Clemmons, D.R. Insulin-like growth factor binding proteins and their role in controlling igf actions. Cytokine Growth Factor Rev. 1997, 8, 45–62.
- Jones, J.I.; Clemmons, D.R. Insulin-like growth factors and their binding proteins: Biological actions. Endocr. Rev. 1995, 16, 3–34.
- Janssen, J.A.; Lamberts, S.W. Is the measurement of free igf-i more indicative than that of total igf-i in the evaluation of the biological activity of the gh/igf-i axis? J. Endocrinol. Invest. 1999, 22, 313–315.
- Philippou, A.; Maridaki, M.; Pneumaticos, S.; Koutsilieris, M. The complexity of the igf1 gene splicing, posttranslational modification and bioactivity. Mol. Med. 2014, 20, 202–214.
- Steele-Perkins, G.; Turner, J.; Edman, J.C.; Hari, J.; Pierce, S.B.; Stover, C.; Rutter, W.J.; Roth, R.A. Expression and characterization of a functional human insulin-like growth factor i receptor. J. Biol. Chem. 1988, 263, 11486–11492.
- LeRoith, D.; Werner, H.; Roberts, C.T. Molecular and Cellular Biology of the Insulin-Like Growth Factors. In Molecular Endocrinology, Basic Concepts and Clinical Correlations; Weintraub, B.D., Ed.; Raven Press: New York, NY, USA, 1995; pp. 181–193.
- Siddle, K.; Urso, B.; Niesler, C.A.; Cope, D.L.; Molina, L.; Surinya, K.H.; Soos, M.A. Specificity in ligand binding and intracellular signalling by insulin and insulin-like growth factor receptors. Biochem. Soc. Trans. 2001, 29, 513–525.
- Froesch, E.; Zenobi, P.; Zapf, J. Metabolic Effects of Insulin-Like Growth Factor-i and Possible Therapeutic Aspects in Diabetes. John Wiley: Chichester, UK, 1993; pp. 109–129.
- Janssen, J.A.; Varewijck, A.J. Igf-ir targeted therapy: Past, present and future. Front. Endocrinol. (Lausanne) 2014, 5, 224.
- Hassan, A.B. Keys to the hidden treasures of the mannose 6-phosphate/insulin-like growth factor 2 receptor. Am. J. Pathol. 2003, 162, 3–6.
- Varewijck, A.J.; Janssen, J.A. Insulin and its analogues and their affinities for the igf1 receptor. Endocr. Relat. Cancer 2012, 19, F63–F75.
- Roberts, C.T., Jr. Control of insulin-like growth factor (igf) action by regulation of igf-i receptor expression. Endocr. J. 1996, 43, S49–S55.
- Rechler, M.M.; Nissley, S.P. The nature and regulation of the receptors for insulin-like growth factors. Annu. Rev. Physiol. 1985, 47, 425–442.
- Ullrich, A.; Gray, A.; Tam, A.W.; Yang-Feng, T.; Tsubokawa, M.; Collins, C.; Henzel, W.; Le Bon, T.; Kathuria, S.; Chen, E. Insulin-like growth factor i receptor primary structure: Comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J. 1986, 5, 2503–2512.
- LeRoith, D.; Werner, H.; Beitner-Johnson, D.; Roberts, C.T., Jr. Molecular and cellular aspects of the insulin-like growth factor i receptor. Endocr. Rev. 1995, 16, 143–163.
- Lopaczynski, W.; Terry, C.; Nissley, P. Autophosphorylation of the insulin-like growth factor i receptor cytoplasmic domain. Biochem. Biophys. Res. Commun. 2000, 279, 955–960.
- Favelyukis, S.; Till, J.H.; Hubbard, S.R.; Miller, W.T. Structure and autoregulation of the insulin-like growth factor 1 receptor kinase. Nat. Struct. Biol. 2001, 8, 1058–1063.
- Myers, M.G., Jr.; Zhang, Y.; Aldaz, G.A.; Grammer, T.; Glasheen, E.M.; Yenush, L.; Wang, L.M.; Sun, X.J.; Blenis, J.; Pierce, J.H. Ymxm motifs and signaling by an insulin receptor substrate 1 molecule without tyrosine phosphorylation sites. Mol. Cell Biol. 1996, 16, 4147–4155.
- White, M.F. Irs proteins and the common path to diabetes. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E413–E422.
- Traub, L.M.; Bonifacino, J.S. Cargo recognition in clathrin-mediated endocytosis. Cold Spring Harb. Perspect. Biol. 2013, 5, a016790.
- Irannejad, R.; Tsvetanova, N.G.; Lobingier, B.T.; von Zastrow, M. Effects of endocytosis on receptor-mediated signaling. Curr. Opin. Cell Biol. 2015, 35, 137–143.
- Yoneyama, Y.; Lanzerstorfer, P.; Niwa, H.; Umehara, T.; Shibano, T.; Yokoyama, S.; Chida, K.; Weghuber, J.; Hakuno, F.; Takahashi, S.I. Irs-1 acts as an endocytic regulator of igf-i receptor to facilitate sustained igf signaling. eLIFE 2018, 7, e32893.
- Takahashi, S.I. Igf research 2016–2018. Growth Horm. Igf Res. 2019, 48–49, 65–69.
- Worrall, C.; Nedelcu, D.; Serly, J.; Suleymanova, N.; Oprea, I.; Girnita, A.; Girnita, L. Novel mechanisms of regulation of igf-1r action: Functional and therapeutic implications. Pediatr. Endocrinol. Rev. 2013, 10, 473–484.
- Vecchione, A.; Marchese, A.; Henry, P.; Rotin, D.; Morrione, A. The grb10/nedd4 complex regulates ligand-induced ubiquitination and stability of the insulin-like growth factor i receptor. Mol. Cell Biol. 2003, 23, 3363–3372.
- Girnita, L.; Girnita, A.; Larsson, O. Mdm2-dependent ubiquitination and degradation of the insulin-like growth factor 1 receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 8247–8252.
- Girnita, L.; Shenoy, S.K.; Sehat, B.; Vasilcanu, R.; Vasilcanu, D.; Girnita, A.; Lefkowitz, R.J.; Larsson, O. Beta-arrestin and mdm2 mediate igf-1 receptor-stimulated erk activation and cell cycle progression. J. Biol. Chem. 2007, 282, 11329–11338.
- Girnita, L.; Shenoy, S.K.; Sehat, B.; Vasilcanu, R.; Girnita, A.; Lefkowitz, R.J.; Larsson, O. -arrestin is crucial for ubiquitination and down-regulation of the insulin-like growth factor-1 receptor by acting as adaptor for the mdm2 e3 ligase. J. Biol. Chem. 2005, 280, 24412–24419.
- Girnita, L.; Takahashi, S.I.; Crudden, C.; Fukushima, T.; Worrall, C.; Furuta, H.; Yoshihara, H.; Hakuno, F.; Girnita, A. Chapter seven - when phosphorylation encounters ubiquitination: A balanced perspective on igf-1r signaling. Prog. Mol. Biol. Transl. Sci. 2016, 141, 277–311.
- De Meyts, P.; Sajid, W.; Palsgaard, J.; Theede, A.-M.; Gaugain, L.; Aladdin, H.; Whittaker, J. Insulin and igf-i Receptor Structure and Binding Proteins. In Mechanisms of Insulin Action; E.Pessin, A.R.S.J., Ed.; Landes Bioscience: Austin, TX, USA, 2007; pp. 1–32.
- Zapf, J.; Schmid, C.; Froesch, E.R. Biological and immunological properties of insulin-like growth factors (igf) i and ii. Clin Endocrinol. Metab. 1984, 13, 3–30.
- Dupont, J.; LeRoith, D. Insulin and insulin-like growth factor i receptors: Similarities and differences in signal transduction. Horm. Res. 2001, 55 (suppl. 2), 22–26.
- Bevan, P. Insulin signalling. J. Cell Sci. 2001, 114, 1429–1430.
- Boucher, J.; Charalambous, M.; Zarse, K.; Mori, M.A.; Kleinridders, A.; Ristow, M.; Ferguson-Smith, A.C.; Kahn, C.R. Insulin and insulin-like growth factor 1 receptors are required for normal expression of imprinted genes. Proc. Natl. Acad. Sci. USA 2014, 111, 14512–14517.
- Boucher, J.; Tseng, Y.H.; Kahn, C.R. Insulin and insulin-like growth factor-1 receptors act as ligand-specific amplitude modulators of a common pathway regulating gene transcription. J. Biol. Chem. 2010, 285, 17235–17245.
- Slaaby, R. Specific insulin/igf1 hybrid receptor activation assay reveals igf1 as a more potent ligand than insulin. Sci. Rep. 2015, 5, 7911.
- Adams, T.E.; Epa, V.C.; Garrett, T.P.; Ward, C.W. Structure and function of the type 1 insulin-like growth factor receptor. Cell Mol. Life Sci. 2000, 57, 1050–1093.
- Mosthaf, L.; Grako, K.; Dull, T.J.; Coussens, L.; Ullrich, A.; McClain, D.A. Functionally distinct insulin receptors generated by tissue-specific alternative splicing. Embo. J. 1990, 9, 2409–2413.
- Slaaby, R.; Schaffer, L.; Lautrup-Larsen, I.; Andersen, A.S.; Shaw, A.C.; Mathiasen, I.S.; Brandt, J. Hybrid receptors formed by insulin receptor (ir) and insulin-like growth factor i receptor (igf-ir) have low insulin and high igf-1 affinity irrespective of the ir splice variant. J. Biol. Chem. 2006, 281, 25869–25874.
- Benyoucef, S.; Surinya, K.H.; Hadaschik, D.; Siddle, K. Characterization of insulin/igf hybrid receptors: Contributions of the insulin receptor l2 and fn1 domains and the alternatively spliced exon 11 sequence to ligand binding and receptor activation. Biochem. J. 2007, 403, 603–613.
- Pandini, G.; Frasca, F.; Mineo, R.; Sciacca, L.; Vigneri, R.; Belfiore, A. Insulin/insulin-like growth factor i hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J. Biol. Chem. 2002, 277, 39684–39695.
- Zhang, H.; Pelzer, A.M.; Kiang, D.T.; Yee, D. Down-regulation of type i insulin-like growth factor receptor increases sensitivity of breast cancer cells to insulin. Cancer Res. 2007, 67, 391–397.
- King, E.R.; Wong, K.K. Insulin-like growth factor: Current concepts and new developments in cancer therapy. Recent Pat. Anticancer Drug Discov. 2012, 7, 14–30.
- Morgillo, F.; Woo, J.K.; Kim, E.S.; Hong, W.K.; Lee, H.Y. Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib. Cancer Res. 2006, 66, 10100–10111.
- van der Veeken, J.; Oliveira, S.; Schiffelers, R.M.; Storm, G.; van Bergen En Henegouwen, P.M.; Roovers, R.C. Crosstalk between epidermal growth factor receptor- and insulin-like growth factor-1 receptor signaling: Implications for cancer therapy. Curr. Cancer Drug Targets 2009, 9, 748–760.
- Guix, M.; Faber, A.C.; Wang, S.E.; Olivares, M.G.; Song, Y.; Qu, S.; Rinehart, C.; Seidel, B.; Yee, D.; Arteaga, C.L. Acquired resistance to egfr tyrosine kinase inhibitors in cancer cells is mediated by loss of igf-binding proteins. J. Clin. Investig. 2008, 118, 2609–2619.
- Liefers-Visser, J.A.L.; Meijering, R.A.M.; Reyners, A.K.L.; van der Zee, A.G.J.; de Jong, S. Igf system targeted therapy: Therapeutic opportunities for ovarian cancer. Cancer Treat. Rev. 2017, 60, 90–99.
- Gee, J.M.; Robertson, J.F.; Gutteridge, E.; Ellis, I.O.; Pinder, S.E.; Rubini, M.; Nicholson, R.I. Epidermal growth factor receptor/her2/insulin-like growth factor receptor signalling and oestrogen receptor activity in clinical breast cancer. Endocr. Relat. Cancer 2005, 12 (suppl. 1), S99–S111.
- Pandini, G.; Mineo, R.; Frasca, F.; Roberts, C.T., Jr.; Marcelli, M.; Vigneri, R.; Belfiore, A. Androgens up-regulate the insulin-like growth factor-i receptor in prostate cancer cells. Cancer Res. 2005, 65, 1849–1857.
- Coppola, D.; Ferber, A.; Miura, M.; Sell, C.; D’Ambrosio, C.; Rubin, R.; Baserga, R. A functional insulin-like growth factor i receptor is required for the mitogenic and transforming activities of the epidermal growth factor receptor. Mol. Cell Biol. 1994, 14, 4588–4595.
- Gan, Y.; Zhang, Y.; Buckels, A.; Paterson, A.J.; Jiang, J.; Clemens, T.L.; Zhang, Z.Y.; Du, K.; Chang, Y.; Frank, S.J. Igf-1r modulation of acute gh-induced stat5 signaling: Role of protein tyrosine phosphatase activity. Mol. Endocrinol. 2013, 27, 1969–1979.
- Tsui, S.; Naik, V.; Hoa, N.; Hwang, C.J.; Afifiyan, N.F.; Sinha Hikim, A.; Gianoukakis, A.G.; Douglas, R.S.; Smith, T.J. Evidence for an association between thyroid-stimulating hormone and insulin-like growth factor 1 receptors: A tale of two antigens implicated in graves’ disease. J. Immunol. 2008, 181, 4397–4405.
- Smith, T.J.; Janssen, J. Insulin-like growth factor-i receptor and thyroid-associated ophthalmopathy. Endocr. Rev. 2019, 40, 236–267.