Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
This entry analyzes the causes and consequences of apoptosis and oxidative stress that occur in mitochondria and cells exposed to the toxic effects of different valent heavy metals (Ag+, Tl+, Hg2+, Cd2+, Pb2+, Al3+, Ga3+, In3+, As3+, Sb3+, Cr6+, and U6+). Experiments with different cells and mitochondria showed that the heavy metals under review induced apoptosis characterized by caspase-3 and -9 activation, Bax and Bcl-2 expression, and mitogen-activated protein kinases.
- heavy metals
- apoptosis
- oxidative stress
- toxic mechanisms
1. Introduction
Early research on heavy metal toxicity. This section is dedicated to a summary of heavy metal toxicity studies that have been performed in previous years. This summary will allow the reader to trace the evolution of the efforts of various researchers on this issue and to understand why the emphasis has shifted from the research of isolated mitochondria to the study of various cellular systems in vitro and in vivo.
Ag(I). It was previously shown that intense rat liver mitochondria (RLM) swelling was induced after the reaction of Ag+ with 12% rapidly reacting SH groups of the inner membrane [1]. Ag+ inhibited Na+, K+-ATPase [2], and mitochondrial malate dehydrogenase from bovine brain [3]. Ag+ increased K+ efflux, coincident with net Ca2+ uptake, and it stimulated the ouabain-sensitive oxygen consumption rate in experiments with rabbit renal cortical tubule suspensions [4][5]. Thiol reagents (n-ethylmaleimide (NEM), dithiothreitol (DTT), and reduced glutathione) reversed these Ag+ effects.
Tl(I). The proximity of the crystal chemical radii of K+ and Tl+ allows the latter to use K+ transport systems to enter the cell [6][7][8]. Na+/K+-ATPase, the Na+-K+-2Cl− cotransporter, and cellular membrane potassium channels are the main systems transporting Tl+ ions into cells [9][10][11][12]. Easy polarizing, compared to K+, allows Tl+ to form stronger chemical bonds with reactive electron donor groups of biological molecules (R-CO2−, R-OPO32−, and R-SH). For this reason, compared to K+, Tl+ has a markedly greater affinity with cellular potassium channels and transporters [10][13]. These features underlie thallium toxicity in particular. The transport of K+ was inhibited by Tl+ in rat myocardial culture cells [14]. Tl+ penetrated the cytoplasm and damaged cellular and mitochondrial membranes in omnifarious body organs [9][15][16][17][18]. Thallium uncoupled oxidative phosphorylation in the mitochondria of mammalian cells [7][19][20][21]. The cytoplasmic concentration of Ca2+ and Na+ increased, while that of K+ fell due to the Tl+ toxic influence on rat hepatocytes and cardiomyocytes [22][23]. Unlike bivalent heavy metals, Tl+ does not have any noticeable effect on either mitochondrial respiratory enzymes (NADH-, succinate-, and malate-dehydrogenases) or mitochondrial thiol groups [9][10][24][25][26]. Here, it must be emphasized that the complexation constants of Tl+ with molecules with vicinal thiol groups turned out to be two orders of magnitude lower than those of ions of toxic heavy metals (Ag+, Hg2+, Cd2+, and Pb2+) [27].
Hg(II). It was previously shown that Hg2+ ions induced oxidative stress that manifested in a decrease in the reduced glutathione concentration and an increase in reactive oxygen species (ROS) or H2O2 production, lipid peroxidation, and state 4 respiration in experiments with energized rat kidney mitochondria (RKM) [28][29][30][31]. At the same time, there was the inner mitochondrial membrane potential (ΔΨmito) decline, Ca2+ release, a respiration decrease in 3 and 3UDNP (2,4-dinitrophenol (DNP)-uncoupled) states, an attenuation of atractyloside-insensitive ADP uptake, and a decrease in both Ca2+ accumulation and mitochondrial ATPase activity (of both basal and Mg2+-activated oligomycin-sensitive components). A Hg2+-induced respiration decrease was detected in experiments with RKM energized by substrates of the inner mitochondrial membrane complexes: CI and CII but not CIV [28]. The above toxic Hg2+ effects were accounted for by a reaction of Hg2+ with CI and CIII, and they could be additionally potentized by Ca2+ followed by the blocking of ruthenium red (RR) or ethylene glycol-bis(β-aminoethyl ether) N,N,N′,N′-tetraacetic acid (EGTA) [30]. Some of these Hg2+ effects were partly prevented by DTT but not cyclosporine A (CsA) or Mg2+. Additionally, Hg2+ activated proton ejection from the matrix of isolated beef heart mitochondria [32]. The H+ ejection and K+ uptake were more substantial in similar research with fluorescein mercuric acetate [32]. It is very interesting that the collapse of ΔΨmito and a decrease in both state 3 respiration and Ca2+ accumulation were found in kidney mitochondria isolated from mercury-poisoned rats [33].
Hg2+ (up to 5 μM) and MeHg (up to 1.5 μM) reduced ΔΨmito in sheep T cells [34]. Hg2+ significantly disturbed the intracellular structure of L929 mouse fibroblasts and inhibited mitochondrial dehydrogenase activity [35]. Experiments with methylmercury hydroxide-exposed rats showed that resistance to Hg2+-induced oxidative stress was supposed to be associated with increasing glutathione (GSH) synthesis in the kidney cortex rather than with an increase in the GSH level itself [36].
Cd(II). Cd2+ has previously been shown to increase the passive permeability of the inner membrane to univalent cations (H+, K+, Na+, and Li+), since the swelling of non-energized RLM and RKM increased in the series of Li+ < Na+ < K+ < NH4+ in experiments with chloride media [37][38][39]. Therewith, Cd2+ built up K+ transport in energized RLM and rat kidney mitochondria (RKM) [37][38][39][40][41][42]. Cd2+ induced mitochondrial swelling, increased state 4 respiration and inorganic phosphate (Pi) uptake, attenuated state 3 and 3UDNP respiration, and induced ΔΨmito decline and Ca2+ efflux in RLM and rat heart mitochondria (RHM) energized by the substrates of mitochondrial complexes (CI, CII, and CIV) [37][38][40][42][43][44][45][46][47]. The short-term Cd2+-induced state 4 increase was eliminated by RR but not by NEM [45][48]. Cd2+-induced mitochondrial uncoupling (state 40 increase) increases in the presence of Pi because Cd2+ stimulates the mitochondrial Pi uptake via the Pi/H+ symporter [45]. Similar potentiating Cd2+-induced effects to these states were found in a buffer with KCl replaced by NaCl but not LiCl [37]. A state 4 respiration increase with proton gradient discharge was found in experiments with RLM in the presence of low Cd2+; however, high Cd2+ induced a short-term increase in respiration, which was followed by a potent respiratory decrease due to the Cd2+ inhibition of CI and CII. However, even high Cd2+ concentrations (more 100 μM) did not inhibit state 4 respiration in RLM energized by a CIV substrate (ascorbate + N,N,N,N-tetramethyl-p-phenylenediamine (TMPD)) [38][43][44][49]. Cd2+ induced the efflux of protons into a sucrose medium containing 10 mM Me+ acetate in the series of Na+ < Li+ < K+ [41]. So, Cd2+ was concluded to inhibit CI–CIII but not CIV. DTT and RR prevented these Cd2+ effects. La3+ inhibited the Cd2+-induced swelling of succinate-energized RLM in a sucrose medium [41]. The chronic administration of low-dose CdCl2 to rats induced oxidative stress that resulted in a hepatic lipid peroxidation increase, hepatic glutathione depletion, and hepatic nuclear DNA damage in liver and brain mitochondria [50].
The Cd2+ inhibition of the membrane-bound succinate dehydrogenase of beef heart was weakened by succinate and malonate [51]. Heavy metals (Hg2+, Cd2+, and Zn2+) with a high affinity for molecular SH groups attenuated Ca, Mg-ATPases’ activity and resulted in oxidative stress disturbing mitochondrial functions in the cells of different organisms [52]. Rod-selective apoptosis resulted in Ca2+ and/or Pb2+ elevation in rat photoreceptor cells [53]. Mitochondrial depolarization, an ATP synthesis decrease, swelling, release, and caspase-9 and caspase-3 activation were observed. These effects (Ca2+ and Pb2+) were additive and blocked completely by CsA [53][54].
Pb(II). Ruthenium red, N3-, and La3+ were previously shown to inhibit the uptake of Pb2+ by isolated rat kidney mitochondria [55]. Ca2+ uptake inhibition, ΔΨmito decline, a swelling and state 40 increase, a state 3 decrease, and a fast release of accumulated Ca2+ were induced by Pb2+ in experiments with RKM, which were energized by CI–CII substrates [56][57][58]. RR prevented the Pb2+-induced ΔΨmito decline, state 3 respiration decrease, and Ca2+ release in RKM energized by succinate but not by NAD-dependent substrates [55][56]. In experiments with bovine heart mitochondria (BHM), energized by CI–CII substrates, Pb2+ inhibited Ca2+ uptake; resulted in mitochondrial swelling and ΔΨmito decline; increased the inner mitochondrial membrane (IMM) passive K+ permeability; and decreased K+ uptake and mitochondrial respiration in 4, 3, 3UCCP, and 3UDNP states [59][60][61]. Some of these Pb2+ effects were blocked by Pi or chlorpromazine [61].
In vivo experiments with rats injected with 203Pb found appreciable lead quantities in the protein fractions but not in the lipid fractions of the inner membrane, the matrix, and the outer membrane of rat liver [62]. Research with rats exposed to Pb(NO3)2 found hepatocyte apoptosis with an activity decrease in the mitochondrial tricarboxylate carrier [63]. Pb2+ and Ca2+ competed for the plasma membrane transport systems and Ca2+-dependent effector mechanisms like calmodulin coupling several enzymes (phosphodiesterase and protein kinases) [64][65]. Pb2+ inhibited Ca2+ uptake in body organ cells and displaced Ca2+ and K+ from isolated mitochondria [64][66]. Pb2+ induced a cell viability decrease and a lipid peroxidation increase in experiments with cultured human fibroblasts [67].
Al(III), Ga(III), and In(III). Early research found that Al3+, some related metals (Sc3+, Ga3+, and In3+), and Be2+ stimulated carbonyl production and Fe2+-initiated lipid and protein oxidation in experiments in vitro with rat brain myelin and synaptic membranes [68]. Al3+ intoxication in vivo was manifested as an increase in lipid peroxidation and myelin fluidity in mouse brain [68]. Ga3+ and In3+ did not show any mitochondrial dehydrogenase inhibition or intracellular structure disruption in experiments with L929 mouse fibroblasts [35]. Experiments with rat brain myelin membranes showed that Fe2+-initiated lipid and protein oxidation decreased in the order of Sc3+, Y3+, La3+ > Al3+, Ga3+, In3+ > Be2+ [68].
As(III). AsO2− markedly inhibited the Ca2+ uptake in RLM energized by a CI substrate (pyruvate) but not succinate [69]. The AsO2−-induced increase in state 40 respiration in succinate-energized RLM was completely inhibited by butylhydroxytoluene (a free radical scavenger) or N,N′-dicyclohexylcarbodiimide (an F1FO-ATPase inhibitor) and partly inhibited by oligomycin [70]. State 3U respiration was completely inhibited in experiments with RLM energized by 2-oxoglutarate but not succinate in the presence of uncouplers (DNP and Cl-CCP) [71]. AsO2− reacting with vicinal dithiols was found to enhance the H+-ATPase proton conductivity in the submitochondrial particles of beef heart mitochondria [72] and to inhibit beta-oxidation in isolated RLM [73]. These results indicate that arsenite significantly inhibits CI but not CII in these mitochondria. On the contrary, AsO2− inhibited mitochondrial succinate dehydrogenase in mouse neuroblastoma N2a cells, but H2AsO4− stimulated it [19]. Glutathione oxidation and the cross-linking of vicinal dithiols by arsenite or phenylarsine oxide (PAO) increased the probability of MPTP opening in RLM [74]. However, NEM eliminated this effect. However, Pi (H2PO4−) or arsenate (H2AsO4−) in Ca2+-loaded RLM induced MPTP opening, which was followed by an increase in H2O2 production, lipid peroxidation, and mitochondrial swelling [75].
Cr(VI). Cr2O72− resulted in lysosomal membrane rupture, reduced glutathione oxidation, increased ROS production and lipid peroxidation, and ΔΨmito decline in isolated rat hepatocytes [76]. Antioxidants, ROS scavengers, and Mn2+ prevented this Cr2O72−-induced cytotoxicity. Mn2+ was supposed to prevent the formation of active Cr(IV) intermediates determining the toxic effects of Cr2O72− [76][77].
2. Modern Research on Heavy Metal Toxicity
Ag(I). Mitochondrial research. Low Ag+ showed a state 4 increase, but high Ag+ resulted in ΔΨmito decline and the complete inhibition of state 3 and state 4 respiration as well as an increase in the inner membrane proton and potassium permeability, ROS production, lipid peroxidation, and swelling in experiments with RLM energized by glutamate or succinate due to the inhibition of mitochondrial CI and CII but not CIV [78][79][80]. Only DTT (a thiol reagent) completely inhibited these Ag+ effects, while MPTP inhibitors (CsA and ADP), ethylenediaminetetraacetic acid (EDTA, a bivalent metal chelator), and RR (a mitochondrial Ca2+ uniporter blocker) had no effect [79][80]. S-15176 but not CsA inhibited Ag+-induced swelling, cytochrome c release, and MPTP opening in succinate-energized RLM [80][81]. Silver nanoparticles (AgNPs) induced MPTP opening and an increase in mitochondrial swelling, state 4 respiration, lipid peroxidation, and ROS production as well as ΔΨmito decline, a state 3 and Ca2+ retention decrease, and a negligible state 3UFCCP decline in experiments with succinate-energized RLM [82][83]. The increase in AgNP-induced swelling was noticeably suppressed by DTT and ethylene glycol-bis(β-aminoethyl ether) N,N,N′,N′-tetraacetic acid (EGTA) but not by MPTP inhibitors (ADP and CsA), and the swelling was additionally accelerated in the presence of RR [82]. AgNP-induced mitochondrial swelling additionally increased in Ca2+-loaded RLM, which was followed by the inhibition of CsA [83].
Ag(I). Cell research. Ag+ induced mitochondrial apoptosis in human Chang liver cells, Ewing’s sarcoma A673 cells, A549 lung cancer cells, human airway smooth muscle, human breast SKBR3 cancer cells, histiocytic lymphoma U-937 cells, human promyelocytic HL-60 cells, human keratinocytes, CHO-K line cells, human bronchial epithelial BEAS-2B cells, human skin fibroblasts, and mouse AML12 hepatocytes [84][85][86][87][88][89][90][91][92][93][94][95][96][97]. The apoptosis manifested alongside decreased cell viability, caspase 3 and 7 activation, pro-apoptotic genes (Bax and Bcl-2) expression, increased metallothionein expression, ATP depletion, and DNA fragmentation. Oxidative stress was also present here as a decline in mitochondrial oxygen consumption, mitochondrial uncoupling, the inhibition of inner mitochondrial membrane complexes I and II (CI and CII), a decrease in reduced glutathione, the loss of ΔΨmito, an increase in ROS production and lipid peroxidation, and cytochrome c release from the mitochondria as well as an increase in cytoplasmic and mitochondrial Ca2+. Some of these Ag+ effects were prevented by thiol reagents (DTT, N-acetylcysteine (NAC), and reduced glutathione).
Silver nanoparticles (AgNPs) have found wide application in both biomedical and consumer products [84]. Silver nanoparticles (AgNPs) and Ag+ with a 1,10-phenanthroline complex induced mitochondrial apoptosis with caspase-3 and caspase-9 activation, decreased cell viability, and disrupted plasma and mitochondrial membrane integrity in experiments with hepatoblastoma HepG2 cells and A549 lung cancer cells [84][92][98]. AgNP-induced mitochondrial oxidative stress manifested as a decrease in reduced glutathione, mitochondrial uncoupling, ΔΨmito decline, a decrease in the antioxidant enzymes’ activity, ATP synthesis stoppage, an increase in membrane leakage, ROS production, lipid peroxidation, and NADPH oxidase activity in experiments with BRL-3A hepatocytes, human keratinocytes HaCaT cells, human peripheral blood mononuclear cells, and mouse AML12 hepatocytes [97][98][99][100][101]. Microscopic studies found that silver-nanoparticle-exposed cells showed an abnormal size, cellular shrinkage, and irregular shape acquisition [99]. The interplay between AgNP-induced mitochondrial fission and mitophagy defects was shown in in vivo and in vitro experiments with human hepatocellular carcinoma HepG2 cells [102]. The features of the AgNP reaction with cellular and mitochondrial enzymes and structures allow for the further use of these particles in different areas, such as anticancer and antimicrobial therapy; the textiles, water treatment, and cosmetics industries; and biomedical research as antimicrobial, antifungal, antiviral, anti-inflammatory, and anti-angiogenic agents [100][101][102][103].
The crystal chemical radii of Ag+ and K+ ions are close in magnitude; therefore, Ag+ can be transported through biological membranes via potassium channels. However, although Ag+ has the highest affinity for the thiol groups of biomolecules and cellular structures, it cannot be classified as the most highly toxic agent. The general explanation is the ability of Ag+ to react with Cl− anions and metallothioneins binding heavy metal ions. So, these interactions markedly decrease the active concentration of Ag+ in the body’s internal environment. Silver nanoparticles are devoid of these shortcomings, and they are able to reach critical thiol groups. Recent comparative studies of AgNPs and Ag+ indicate this circumstance [104][105][106]. It was AgNPs that maximally reached and integrated into cellular structures, exerting a more substantial toxic effect on cells than Ag+. Understanding the factors determining the toxicity of AgNPs is very important for biomedical applications, particularly in cancer therapy. In this regard, modern studies of silver nanoparticles are very relevant in terms of the search for new anticancer and antimicrobial drugs. The Ag+ ions that overcome these barriers damage DNA, inhibit respiratory chain complexes, and induce MPTP opening, which are accompanied by the calcium overload of mitochondria. Together, Ag+ ions cause oxidative stress (Figure 1), which is accompanied by a decrease in ATP synthesis and the development of apoptotic processes (Figure 2) leading to cell death. Undoubtedly, an excessive accumulation of silver in the body can contribute to developing diseases associated with metabolic disorders and lead to the degradation of body systems and the development of oncology.
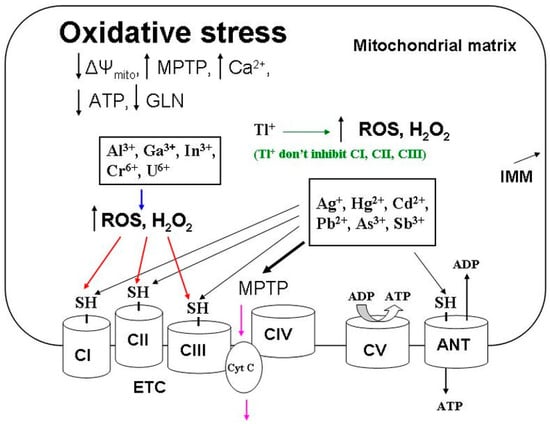
Figure 1. General elements for toxic mechanisms of inducing mitochondrial oxidative stress via heavy metals. Heavy metals (Ag+, Hg2+, Cd2+, Pb2+, As3+, and Sb3+) induce oxidative stress by blocking respiratory complexes with MPTP opening due to interacting with the thiol groups shown in the picture. Other metals (Tl+, Al3+, Ga3+, In3+, Cr6+, and U6+) inactivate these thiol groups by indirectly oxidizing them due to activation of the production of oxygen radicals (ROS and H2O2). Black arrows show the metal’s reaction with thiol groups (SH-). The MPTP induction by heavy metals is shown by the bold arrow. The induction of ROS and H2O2 is marked with a blue arrow. The ROS-induced oxidation of thiol groups is shown by red arrows. Pink arrows show the cytochrome c release into the intermembrane space due to the MPTP opening. Abbreviations: ADP, adenosine diphosphate; ANT, adenine nucleotide translocase; ATP, adenosine triphosphate; CI–CV, inner mitochondrial membrane complexes I–V; Cyt C, cytochrome C; ETC, electron transport chain; GSH, reduced glutathione; IMM, inner mitochondrial membrane; MPTP, mitochondrial permeability transition pore; ROS, reactive oxygen species; SH-, molecular thiol groups; ΔΨmito, inner membrane potential.
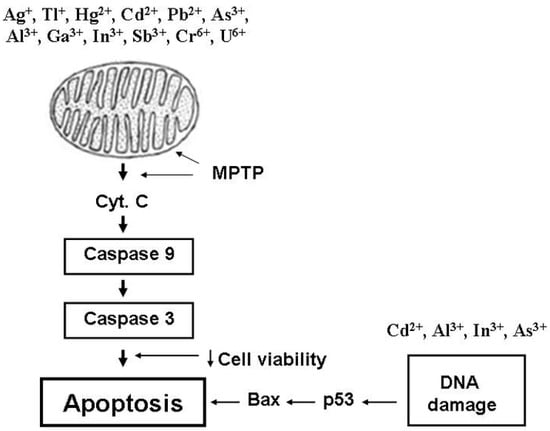
Figure 2. General links in mitochondria-dependent pathways of intracellular apoptosis induction by different-valence heavy metals. Heavy metals (Ag+, Tl+, Hg2+, Cd2+, Pb2+, As3+, Sb3+, Al3+, Ga3+, In3+, Cr6+, and U6+) induce mitochondrial permeability transition pore (MPTP) opening, which is accompanied by cytochrome c release into the intermembrane space. As a result of this process, caspases-3 and -9 are activated and trigger apoptosis induction. Some metals (Cd2+, As3+, Al3+, and In3+) damage DNA, resulting in bax and p53 activation that induces apoptotic processes. Arrows show MPTP induction. Bold vertical arrows show the sequence of events associated with mitochondria during the induction of intracellular apoptosis by heavy metals. Apoptosis induction pathways associated with DNA damage are indicated by horizontal arrows. Abbreviations: Cyt. C, cytochrome C; MPTP, mitochondrial permeability transition pore.
Tl(I). Mitochondrial research. The inner mitochondrial membrane turned out to be noticeably permeable to Tl+ ions [107][108]. Excess Tl+ ions are removed from mitochondria when using a K+/H+ exchanger, which is followed by the pumping out of incoming protons from the matrix [109]. In addition, Tl+ does not inhibit mitochondrial respiratory enzymes [9][10][24][25]. This is why Tl+ increased state 4 respiration and did not affect state 3 or 3UDNP respiration in energized RLM [9][24][25][109][110][111][112]. As a similarly toxic heavy metal, Tl+ increased the passive permeability of the IMM to univalent cations (K+, H+, and Na+), and that was shown in experiments with non-energized RLM injected into a nitrate medium containing a mixture of nitrates of Tl+ and one of the univalent cations [109]. Energized RLM added to the nitrate medium showed an increase in swelling and state 4 respiration as well as some decline in state 3 and 3UDNP respiration and the inner mitochondrial membrane potential (ΔΨmito) [109].
It has been previously postulated that thallium(I) toxicity is due to the ability of Tl+ to easily penetrate the IMM and uncouple oxidative phosphorylation [24][107]. Tl+ ions transport into energized mitochondria using both the mitochondrial ATP-sensitive potassium channel (mitoKATP) and the mitochondrial BK-type Ca2+-activated potassium channel (mitoKCa) [113][114]. Massive mitochondrial swelling was found in in vivo experiments using thallium(I) salts [15][25][115]. One reason for this swelling may be MPTP opening in the inner mitochondrial membrane. Such phenomena as mitochondrial swelling, state 3 and 3UDNP decrease, and ΔΨmito decline were markedly increased in experiments with energized calcium-loaded mitochondria [116][117]. These effects were completely eliminated or markedly attenuated by MPTP inhibitors (ADP, CsA, bongkrekic acid (BKA), and NEM), mitochondrial Ca2+ uniporter blockers (RR, Y3+, La3+, Sr2+, and Mn2+), or a Ca2+- chelator (EGTA) [110][116][118][119][120]. Tl+-induced MPTP opening is possible only in the case of calcium-loaded mitochondria, while such loading is not required in similar experiments with heavy bivalent metals. The inhibition of mitoKATP and mitoKCa decreased the matrix calcium retention and accelerated the Tl+-induced MPTP opening in calcium-loaded rat liver mitochondria [121]. The substrate specificity effect [122] was found in succinate-energized mitochondria, which had more resistance to calcium loading than those energized by CI substrates [116]. Tl+-induced MPTP opening is dependent on the adenine nucleotide translocase (ANT) conformation [119]. The relationship between the ANT conformation and MPTP opening in the inner membrane was substantiated previously in detail [123][124]. The pore-opening phenomena were more noticeable in fixing the ANT c-conformation induced by thiol reagents (phenylarsine oxide (PAO), 4,4′-diisothiocyanostilbene-2,2′-disulfonate (DIDS), and high NEM), thiol oxidizers (tert-butyl hydroperoxide (tBHP), diamide (Diam)), and carboxyatractyloside (CAT) [119][125]. On the contrary, the manifestation of these phenomena noticeably decreased or completely inhibited the stabilization of the ANT m-conformation induced by reagents (ADP, low NEM, BKA, and eosin-5-maleimide (EMA)) and low mersalyl [9][119][126]. Additionally, Tl+-induced MPTP opening was recently found to be dependent on the activity of cysteine and lysine residues in the inner membrane proteins [126][127].
Tl(I). Cell research. Apoptosis, membrane integrity disruption, MPTP opening, Na+/K+-ATPase inhibition, glutathione depletion, ΔΨmito decline, cell viability loss, cytochrome c release, membrane fluidity rise, ATP depletion from mitochondria, and an increase in lipid peroxidation, ROS production, and H2O2 synthesis were induced by Tl+ in experiments with isolated rat hepatocytes, isolated synaptosomal mitochondrial fractions of P2, PC12, and HN9.10e neuronal cells, C6 glioma and Jurkat T cells [9][110][128][129][130][131][132][133][134][135][136][137]. Tl+-induced apoptosis in PC12 cells was later shown to be associated with the activation of the MAPK-dependent cascade of protein kinases: extracellular signal-regulated kinase (ERK)1/2, c-jun N-terminal kinase (JNK), p38, and p53 of the Bcl-2 family [133]. Experiments with TlCl-treated rat hepatocytes found increased cytoplasmic concentrations of Ca2+ and Na+ but decreased K+ concentrations [22]. MPTP inhibitors (CsA and carnitine) visibly reduced the cytotoxic effects of Tl+ [131][132]. Thallium(I)-induced metabolic disturbances are situations in which Tl+ inhibits Mg2+-regulated and K+-dependent pyruvate kinases, disrupts glycolysis processes, and interferes with the Krebs cycle [7][9][10][12][13].
Tl+ and K+ cations (as well as Ag+) have similar crystal chemical radii. For this reason, Tl+ ions enter cells through potassium channels. However, Tl+ (unlike Ag+ and other heavy metals) has a minimal affinity for thiol groups. For this reason, Tl+ really does not bind to intracellular metallothioneins and quickly penetrates cells, disrupting potassium-dependent cell processes. This distinguishing Tl feature is especially true of the toxic effects of Tl+ ions on neurons, cardiomyocytes, and kidney cells. With a more prolonged exposure to the body, Tl+ reduces the concentration of reduced glutathione while increasing ROS production in cells. Oxidative stress (Figure 1) and apoptotic processes (Figure 2) develop due to damage to cellular and mitochondrial membranes, decreased ATP synthesis, and the subsequent induced MPTP opening and calcium overload of cells and mitochondria. Such features of the chemical behavior of Tl+ do not allow this metal to be rapidly removed from the body. Future research should focus on finding effective Tl+ binding reagents for the subsequent elimination of this metal from the body to treat thallium-induced damage to the nervous system, kidneys, and hairline [9].
Hg(II). Mitochondrial research. Hg2+ was shown to induce oxidative stress in experiments with RLM energized by CI and CII substrates [42][138][139][140]. There was a state 3 and 3UDNP respiration decrease, ΔΨmito decline, mitochondrial swelling, a state 4 respiration increase, a mitochondrial membrane fluidity increase, cytochrome c release, and MPTP-induced mitochondrial ATP depletion. So, Hg2+ to Cys binding mediates multiple Hg2+ toxic impacts, especially the inhibition of enzymes and other proteins containing free Cys residues causing oxidative stress [141]. These unfavorable effects were prevented by MPTP inhibitors (ADP, CsA, Mg2+, and BKA) and Ru360 (an MCU blocker). F1FO-ATPase activity was promoted by micromolar Hg2+, and respiration in state 4 was inhibited in swine heart mitochondria energized by NADH but not succinate [142]. Hg2+ and MeHg significantly declined mitochondrial viability and increased the H2O2 and ROS production, lipid peroxidation, and glutathione oxidation in mouse brain mitochondria [143]. Quercetin and catalase prevented these effects of Hg2+. Hg2+ resulted in swelling, a Ca2+ efflux, and an ROS production increase in experiments with RKM [144]. The calcium load of these mitochondria additionally increased swelling and induced ΔΨmito decline. Tamoxifen and CsA inhibited these in vitro effects of Hg2+. Ca2+ uptake and Ca2+-induced ΔΨmito decline were inhibited in mitochondria isolated from the kidneys of rats injected with mercury chloride plus tamoxifen [144]. Tamoxifen was concluded to be an inhibitor of Hg2+-induced MPTP [144].
Hg(II). Cell research. MeHg similarly to Hg2+ induces mitochondrial apoptosis and oxidative stress, mediating an increase in ROS production, a decrease in cell antioxidant defense, and cytochrome c release from mitochondria [145][146]. Hg2+ induced apoptosis with caspase-3 activation, a cell viability decrease, mitochondrial integrity decline, and intracellular ATP depletion. There was oxidative stress manifested as a decline in ΔΨmito, cytochrome c release, an increase in ROS generation, an increase in lipid peroxidation, a decrease in ATP and reduced glutathione, and a decrease in thioredoxin reductase and glutathione peroxidase activity in human hepatoma HepG2 cells, human neuroblastoma SH-SY5Y cells, human gingival fibroblasts, hamster pancreatic HIT-T15 β-cells, normal rat kidney cells, rat ascites hepatoma AS-30D cells, and human T cells and leukocytes [147][148][149][150][151][152][153][154][155][156]. N-acetylcysteine reversed some of these effects of Hg2+. Experiments with Hg2+- and MeHg-treated fibroblast zebrafish ZF4 cells found a decrease in ATP production and mitochondrial respiration in the basal, 3, and 3UFCCP states [157]. However, the proton leak and non-mitochondrial respiration were not changed there [157]. Hg2+ induced a cell viability decrease, ΔΨmito decline, and an ROS production increase in experiments with five Tetrahymena species; however, more ingested MeHg disrupted the membrane integrity [158].
The Hg2+- and MeHg-induced apoptosis in HepG2 cells can be due to Hg2+, and MeHg inhibited the respiration, increased the permeability, and blocked the essential thiol proteins of mitochondrial membranes [159]. The inner membrane protein thiols were dose-dependently decreased by Hg2+ in swine heart mitochondria [142]. However, these Hg2+-induced effects were reversed by DTT due to the latter’s interaction with the Cys residues of respiratory complexes [142]. A Fenton-type reaction may be the cause of the Hg2+-induced oxidative stress and ROS production in NRK-52E cells due to the binding of Hg2+ to intracellular thiols (proteins or glutathione) [160]. Hg2+ increased free Ca2+ levels in canine kidney cells due to both extracellular Ca2+ influx (protein kinase C-regulated) and the phospholipase C-activated Ca2+ release from endoplasmic reticulum (ER) [161]. MeHg-induced neurotoxicity in primary rat cortical neurons stimulated Ca2+ release, increasing from the ER simultaneously with MPTP opening and ΔΨmito decline [162].
Hg2+ penetrates cells and mitochondria via calcium-transporting mechanisms. Hg2+ ions induce oxidative stress (Figure 1) and apoptotic processes (Figure 2) in cells and mitochondria, having a noticeable affinity for thiol groups. This stress manifests in a marked decrease in CI and CII activities, a decline in ΔΨmito, the induction of MPTP opening, and a decrease in ATP and GSH synthesis. At the same time, the calcium overload of cells and mitochondria sharply increases, ROS production increases, and the reduced GSH concentration decreases. At higher levels, Hg2+ damages renal nephrons. The main toxic manifestations in this case can be considered the induction of MPTP opening and the blocking of critical thiol groups by Hg2+ ions because the MPTP inhibitors (ADP, CsA, and tamoxifen) and reducers of these groups (DTT and NAC), along with calcium transport blockers (RR and Ru360), eliminated the toxic effects of Hg2+.
Cd(II). Mitochondrial research. Studies of the cadmium effects on various characteristics of isolated mitochondria were conducted. Cd2+ in succinate-energized RLM induced ΔΨmito decline; iron mobilization; a decrease in state 3 and 3UDNP respiration, ATP content, potential dependent 137Cs+ uptake, and protein thiol levels; and an increase in state 40 respiration, IMM proton permeability, and H2O2 production; but Cd2+ slowly affected RLM succinate dehydrogenase activity [48][138][140][163][164]. Cd2+ (similarly Hg2+) increased the inner membrane passive permeability to H+ and K+ ions and showed ΔΨmito collapse, a decrease in 3UDNP respiration, and an increase in K+ mitochondrial uptake and state 40 respiration in RHM energized by CI and CII substrates [42][46][47]. Cd2+ resulted in respiration inhibition in 3 and 3UFCCP states, ΔΨmito decline, cytochrome c release, Ca2+ uptake inhibition, and a Ca2+ release and H2O2 generation increase in isolated rat kidney mitochondria [39][165][166]. DTT and CsA prevented the Cd2+-induced Ca2+ efflux and ΔΨmito decline in energized rat kidney mitochondria [167]. Quinine inhibited the contraction of succinate-energized RKM preswollen in a KCl medium containing Cd2+ [39].
After reacting with the outer and inner thiol groups of the inner membrane, Cd2+ and Hg2+ then penetrate into mitochondria and cause the release of cytochrome c, the opening of MPTP, and the inhibition of CI and CII [42]. After penetrating into the matrix through the mitochondrial Ca2+ uniporter, Cd2+ was assumed to induce mitochondria osmotic swelling and the cytochrome c release due to the activation of aquaporin H2O channels because mitoplast swelling was blocked by Ag+ (a potent aquaporin blocker) [168]. The Cd2+-induced swelling, ΔΨmito decline, state 40 increase, IMM fluidity increase, and lipid peroxidation were decreased in the presence of MPTP inhibitors (ADP, CsA, Mg2+, DTT, NEM, RR, and Ru360) in experiments with succinate-energized RLM [39][44][48][140][169]. Moreover, the maximum effect was achieved with the simultaneous presence of some of these inhibitors in salt media containing NH4NO3, KCl, or sucrose [44]. Atractyloside prevented the contraction of succinate-energized RLM preswollen in a NH4NO3 medium containing 15 µM Cd2+, oligomycin, and rotenone [44]. CsA partly inhibited the Cd2+-induced state 40 increase and state 3UDNP decrease in succinate-energized RLM [44]. Thus, Cd2+-induced mitochondrial dysfunction appears to be associated with the opening of ADP- and CsA-dependent MPT pores in the inner membrane [44]. Cd2+ resulted in MPTP opening, ΔΨmito decline, cytochrome c release, decreased state 3 respiration, and increased state 40 respiration. These Cd2+ effects were completely suppressed by Bcl-xL, RR, and BKA but not by CsA in experiments with succinate-energized mouse liver mitochondria [170]. This Cd2+-induced MPTP opening was supposed to be the reason for the Cd2+ interaction with ANT thiol groups [170].
Mg2+ with the ADP inhibition of the state 40 increase, induced by high Cd2+ with RR, was not changed in experiments with succinate-energized RLM added to a K-acetate medium containing both a mitoKATP inducer (diazoxide) and the channel inhibitors (5-hydroxydecanoate (5-HD) and glibenclamide) [140]. However, the Cd2+-induced swelling of RLM-energized glutamate with malate increased in a KCl medium containing the mitoKATP inhibitor ATP. This effect was slightly prevented by diazoxide and, in contrast, was markedly potentiated by 5-HD [140]. So, one can conclude that the above inhibition of the Cd2+-induced state 40 increase is probably due to the mitoK+ uniporter blocking by ATP with Mg2+ but not the MPTP opening. On the contrary, the Cd2+-induced swelling increase in the KCl medium containing mitoKATP inhibitors (ATP and 5-HD) is probably due to the greater MPTP opening when the mitoKATP channels are closed. This result is in good agreement with the previous assumption that the probability of the MPT pore opening in the inner membrane increases when the mitoKATP channels are closed [171][172][173].
Research on energized RLM found that the state 40 respiration increase and the state 3UDNP respiration decrease induced by Cd2+ with Ca2+ were visibly eliminated in the presence of ADP with CsA [163][174]. However, Cd2+-induced state 40 respiration increased even more after the administration of DTT but not EGTA, which was injected after high Cd2+ [138][163][174]. Ca2+ with Pi or NEM alone markedly increased the Cd2+-induced passive proton permeability of the inner membrane. In this case, preswollen RLM (after the succinate addition) showed some contraction in a NH4NO3 medium containing low NEM (10–100 μM) or Pi alone but not Ca2+ with/without Pi or high NEM (1 mM). This contraction became much more potent after the DTT addition to the medium containing NEM with rotenone, and the succinate-energized mitochondria visibly contracted (like the control Cd2+-free experiments), even in the presence of 1 mM NEM (except for in the experiments where Cd2+, Ca2+, and Pi were all present) [44][163][174].
Cd(II). Cell research. Cd2+ transports into cells by using ion channels, special carriers, Ca2+/calmodlin-dependent protein kinase II (CaMK-II), and ATP-hydrolyzing pumps, whereas Cd2+–protein complexes penetrate the membrane through receptor-mediated endocytosis of the cadmium–metallothionein complexes but not N- and L-type Ca2+ channels, which are closed at the resting membrane potential [175][176][177]. In addition, the mitochondrial Ca2+ uniporter is involved in the Cd2+ transport into mitochondria [175].
Being a physiological secondary messenger, Ca2+ and ROS control Ca2+-dependent and redox-sensitive molecular processes that determine cell function and fate. Ca2+ and ROS are known to activate cell death effectors (caspases, ceramides, calpains, and p38), to irreversibly damage mitochondria and the endoplasmic reticulum, and to modulate biosynthesis metallothioneins and Bcl-2 proteins [178]. It is the Ca2+ and ROS increase that is directly affected by the Cd2+ impact in experiments in vitro and in vivo [178]. Cd2+ produced an increase in the cytoplasm Ca2+ concentration in canine kidney cells, rat primary astrocytes, rat primary cerebral cortical neurons, yeast cells, mouse skin fibroblasts, and rat hepatocytes [179][180][181][182][183][184]. Cd2+ induced the release of the stored Ca2+ from the endoplasmic reticulum and mitochondria in canine kidney cells and NIH 3T3 cells [179][185]. Cd2+ decreased agonist-induced ER calcium signals and sarcoplasmic–ER calcium ATPases activity in NIH 3T3 cells [185].
Caspases (cysteine proteases), ceramides (cell membrane lipids), and calpains (Ca2+-dependent proteases) are known to play an important role in Cd2+-induced apoptosis in rat kidney proximal tubule cells and to be related directly to Cd2+-induced nephrotoxicity in vivo [186]. Cd2+ induced apoptosis involving the activation of caspase-9, caspase-3 and mitogen-activated protein kinases (ERK, JNK, p53, and p38) as well as DNA fragmentation in rat C6 glioma cells, HEK293 cells, Vero cells, rat ascites hepatoma AS-30D cells, human lung cancer A549 cells, hamster ovary CHO-9 cells, PC12 cells, rat and mouse cochlea HEI-OC1 cells, rat primary astrocytes, rat primary cerebral cortical neurons, U-937 human promonocytic cells, mouse skin fibroblasts, NIH 3T3 cells, yeast cells, rat kidney proximal tubule cells, brain newborn rat oligodendrocytes, rat hepatocytes, mouse skin epidermal JB6 Cl41 cells, AS-30D ascites hepatoma cells, and human hepatocellular carcinoma HepG2 cells [140][152][153][155][156][168][180][181][182][184][185][187][188][189][190][191][192][193][194][195][196][197][198][199][200][201][202]. This Cd2+-induced apoptosis was accompanied by oxidative stress and mitochondrial injury, which manifested as a cell viability reduction, CI–CIII dysfunction, ATP depletion, ΔΨmito decline, matrix Ca2+ load, a reduced glutathione decrease, cytochrome c release, an ROS and H2O2 production increase, and lipid peroxidation as well as mitochondrial damage and swelling. Cd2+-induced oxidative stress is affected by this metal interaction with sulfhydryl groups of mitochondrial critical molecules [203]. Na+/K+-ATPase, Ca2+/Mg2+-ATPase, and other transport proteins can be damaged by the Cd2+-induced ROS increase in rat kidney proximal tubule cells and rat primary cerebral cortical neurons [184][193]. Cd2+-induced actin disruption and the activation of Ca2+/calmodulin-dependent protein kinase II preceded the acceleration of apoptotic mouse and rat mesangial cell death involving actin-dependent mitochondrial structure changes, ROS production, and p38 activation [204][205]. Experiments with rats exposed to Cd acetate found Cd2+-induced rat liver oxidative stress manifesting as decreased superoxide dismutase (SOD) activity and GSH content as well as increased catalase activity and thiobarbituric acid content [206].
Cd2+ in normal human lung MRC-5 fibroblasts caused caspase-independent apoptosis mediated by ROS scavengers (NAC and mannitol) to indicate the crucial role of ROS in apoptogenic Cd-activated apoptosis. The Cd2+-induced apoptosis was partially or completely abrogated by CI and CV inhibitors (rotenone and oligomycin A) or MPTP inhibitors (CsA and aristolochic acid) [207].
Cd2+ and Hg2+ were known to induce metallothionein synthesis and alter zinc and copper homeostasis in the liver and kidney tissue of male albino mice [208]. EGTA and polyphenolic compounds (ellagic acid and quercetin) effectively reduced the Cd2+ toxic effects on rat primary astrocytes and goat sperm [180][209]. The Cd2+-induced Ca2+ release in renal tubular cells was inhibited by thapsigargin (an ER Ca2+pump inhibitor) and carbonyl cyanide m-chlorophenyl hydrazone (CCCP, a mitochondrial uncoupler) [179]. Mn2+ prevented the Cd2+-induced calcium increase in cultured cerebellar granule neurons and dose-dependent death rate [210]. Zn2+ inhibited the intracellular Cd2+ bioaccumulation in cultured rat cortical neurons and RAW 264.7 macrophages [211][212]. Zn2+ prevented Cd2+-induced necrosis and apoptosis in PC12 cells, bovine kidney epithelial cells, and rabbit proximal tubule RP1 cells [213][214][215]. Suppressed mitochondrial apoptosis and a decrease in Cd2+-induced ROS production and cytochrome c release, as well as the downregulation of the Bax proapoptotic protein and a lowering of caspase 9 expression, were observed. The Cd2+ effects on rat ascites hepatoma AS-30D cells and PC12 cells were partly attenuated by MPTP inhibitors (CsA and BKA), respiratory CIII inhibitors (stigmatellin and antimycin A), and antioxidants (N-acetylcysteine and butylhydroxytoluene) [153][199]. Ru360 blocked Cd2+-induced apoptosis in rat kidney proximal tubule cells, but MPTP inhibitors (CsA and BKA) had no effect [168][193]. Reduced glutathione and catalase markedly prevented Cd2+-induced DNA fragmentation in rat C6 glioma cells, suggesting the participation of H2O2 production in these apoptotic processes [188]. Cd2+ cytotoxicity was regulated by intracellular glutathione levels in newborn rat brain oligodendrocytes and rat kidney proximal tubule cells [192][194]. Antioxidants (N-acetyl-l-cysteine and ebselen) noticeably decreased Cd2+ toxicity on rat and mouse cochlea HEI-OC1 cells [191]. Glutathione S-transferase P1 overexpression greatly reduced the Cd2+ effects on HEK293 cells [189]. Apoptosis inhibitors (cycloheximide and dactinomycin), a caspase 3 inhibitor, MPTP inhibitors (CsA, carnitine, and trifluoperazine), ROS scavengers (dimethyl sulfoxide (DMSO), mannitol, catalase, and SOD), a Na+/H+ exchanger inhibitor (5-(N,N-dimethyl)-amiloride), and 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA, a specific intracellular Ca2+ chelator) prevented Cd2+-induced cytotoxicity and ΔΨmito decline in rat hepatocytes and rat cerebral cortical neurons [183][184][195][197]. NO prevented Cd2+-induced apoptosis, caspase-3 activation, and lipid peroxidation in rat anterior pituitary cells [216].
Cd2+ actively interacts with the IMM thiol groups of cells and mitochondria. The result of this interaction is the induction of apoptosis (Figure 2) and oxidative stress (Figure 1) manifesting in increased ion transport through IMM, the inhibition of mitochondrial respiratory complexes (CI–CIII), a decrease in ATP synthesis, the calcium overload of mitochondria, an increase in ROS production, a decline in ΔΨmito, and the induction of the MPTP opening. At a higher body level, Cd2+ damages target organ cells (nervous tissue and liver). Considering the closeness of Cd2+ and Ca2+ radii, it should be noted that the toxic effects of Cd2+ increase significantly under conditions of the calcium overload of cells and mitochondria. The above Cd2+ effects, regardless of the presence of calcium, were markedly attenuated in the presence of calcium transport blockers and competitors (RR, Ru360, Sr2+, and Mn2+), MPTP inhibitors (CsA and BKA), and thiol-reducing agents (NAC and DTT). Therefore, the main three toxic Cd2+ manifestations leading to oxidative stress and apoptosis can be considered the Cd2+-induced calcium overload of mitochondria, the blocking of critical thiol groups, and the opening of MPTP. Cadmium toxicity processes can involve mitochondrial s-glutathionylation. Cd2+ disturbed the redox balance of mitochondria due to an ROS production increase, which enhanced the S-glutathionylation of mitofusin 2 and damaged the membranes binding ER with subsequent neuronal necroptosis [217].
Pb(II). Mitochondrial research. Experiments with mitochondria showed that Pb2+-induced mitochondrial oxidative stress manifested in the inhibition of CII–CIV as well as in an increase in ROS production and lipid peroxidation, ATP consumption, and glutathione oxidation [218][219]. In parallel, Pb2+ decreased the activity of mitochondrial antioxidant system enzymes (GPX, SOD, catalase, and GSH) [219]. At the same time, Pb2+ induced MPTP opening that manifested in RLM swelling, a state 3 respiration decrease, ΔΨmito decline, an IMM fluidity decrease, proton influx in the matrix, and K+ and cytochrome c release. The Pb2+-induced swelling visibly declined in the presence of known MPTP inhibitors (CsA and ADP), chelators (EGTA and EDTA), and a mitochondrial Ca2+ uniporter inhibitor (RR) [66][218][219].
Pb(II). Cell research. Pb2+ induced apoptosis, including the activation of caspase-3 and the expression of the apoptotic-inducing factors (Bax, Bcl2, and Bcl-2), MPTP opening, and oxidative stress that manifested as mitochondrial function oxidative damage, cytochrome c release, and a decrease in cell viability, reduced glutathione, and ATP production as well as an ROS production increase, ΔΨmito decline, Ca2+ release from the matrix, and a decline in the mitochondrial Ca2+ uniporter expression in human neuroblastoma SH-SY5Y cells, rat proximal tubular cells, Siberian tiger fibroblasts, rat insulinoma β-cells, PC12 cells, and adult rat hepatic stem cells [220][221][222][223][224][225][226][227]. These effects of Pb2+ were eliminated by MPTP inhibitors (CsA, DIDS, and BKA), which may indicate the participation of Cyp-D and ANT in Pb2+-induced MPTP opening. It was concluded that the mitochondrial Ca2+ influx regulation in neurons is mediated by the Pb2+-induced oxidative stress response.
The mechanism of Pb2+ neurotoxicity is associated with this cation ability to replace or compete with important biogenic cations (Ca2+, Fe2+, and Zn2+) [228]. Pb2+-induced oxidative stress manifested in mitochondrial uncoupling; the inhibition of CI and CIII; a decrease in the matrix reduced glutathione; ΔΨmito; mitochondrial O2 consumption; an increase in the IMM ion permeability, ROS production, and lipid peroxidation; and a decrease in cytoplasm ATP due to an energy consumption imbalance [228]. All these toxic Pb2+ effects led to the displacement of Ca2+ from intracellular compartments, the opening of MPTP, and the induction of apoptotic and necrotic changes in neurons and cells [228]. Pb2+ induced a reduction in glucose-stimulated insulin secretion in experiments with rat insulinoma β-cells [225].
The toxic actions of Cd2+, Hg2+, and Pb2+ were studied in vitro on pancreatic β-cells from CD-1 mice [229]. These metals resulted in oxidative stress manifesting as a decrease in ΔΨmito, ATP production, CI–CIV activity, and oxygen consumption rates with a parallel increase in cell lactate production, mitochondrial succinate-supported swelling, and IMM permeability to K+ and H+ ions as well as an increase in membrane fluidity and a decrease in saturated/unsaturated fatty acid ratios [229]. Pb2+ damaged liver mitochondrial and nuclei structures and activated mitochondrial apoptosis through caspase-3 and caspase-9 signaling pathways in experiments with Pb-poisoned chickens [230]. The independence of the toxic mechanisms of Cd and Pb was proven in experiments with rats exposed to Cd acetate [206]. Pb2+ alone resulted in a decrease in liver superoxide dismutase (SOD) activity; however, catalase activity and glutathione (GSH) or thiobarbituric acid content were unchanged. Cd2+ with/without Pb2+ resulted in rat liver oxidative stress because a decrease in SOD activity and GSH content and an increase in catalase activity and thiobarbituric acid content were found.
A distinctive feature of Pb2+ ions is their ability to compete with Ca2+ for calcium transport systems of the plasma membrane. However, by entering the mitochondria, Pb2+ inhibits Ca2+ uptake and displaces Ca2+ and K+ from the matrix. The main targets of Pb2+ are the cells of the nervous system, pancreas, and liver. Given the ability of lead to displace calcium from cells, osteoblasts may be another reason for Pb2+ toxic effects leading to bone demineralization. After penetrating mitochondria, Pb2+ induces oxidative stress (Figure 1) and apoptotic processes (Figure 2), interacting with thiol groups and displacing essential biogenic cations (Ca2+, Fe2+, and Zn2+) from mitochondrial structures. Such oxidative stress is characterized by decreased CII–CIV activity, MPTP opening, ΔΨmito decline, decreased ATP consumption, increased ROS production and lipid peroxidation, and glutathione oxidation, reducing the activity of mitochondrial antioxidant system enzymes. That is why Pb2+ toxicity visibly declines in the presence of known MPTP inhibitors (ADP, CsA, BKA, and DIDS), chelators (EGTA and EDTA), antioxidants (catalase), and a mitochondrial Ca2+ uniporter inhibitor (RR). Since lead enters the environment due to various technological processes, it has become essential to develop ways to protect the body from the toxic manifestations of lead compounds and remove them from the body. Pb2+ can accumulate in astrocytes and have a pronounced poisonous effect, disrupting glutamate metabolism [231]. Additionally, the activation of S-glutathionylation processes was found because of the enhanced expression of glutathione–protein complexes after Pb treatment.
The accumulation of heavy metals (Hg, Cd, Pb, Cr, U, and As) in living organisms leads to toxic damage to different organs and tissues. These heavy metals disturb various biological functions, including proliferation, differentiation, damage repair, and apoptosis induction [232][233][234]. At the same time, the mechanisms of toxicity induction often have common patterns directly related to the occurrence of mitochondrial oxidative stress. An ROS production increase, antioxidant defense reduction, enzyme inactivation, MPTP opening, calcium overload, and ΔΨmito decline belong to these patterns. The toxic effects of these metals are multilevel and may play a role in the development of cancer, autoimmune diseases, diabetes, toxicogenomic disorders, Parkinson’s disease, and other metabolic disorders.
Al(III), Ga(III), and In(III). Mitochondrial research. Al3+ decreases the Ca2+ uptake in ER, induces the release of Ca2+ from the matrix, and potently inhibits Ca2+-ATPase activity in RLM that can result in intracellular calcium overload [235]. Aluminum nanoparticles (AlNPs), more so than Al3+, induced oxidative stress that manifested as an increase in mitochondrial swelling, ROS production, and lipid peroxidation, as well as CIII inhibition, ΔΨmito decline, and cytochrome c release, in experiments with isolated rat brain mitochondria [236]. Al3+ plus tyramine induced MPTP opening in Ca2+-loaded RLM that manifested as mitochondrial swelling, an ROS and H2O2 production increase, mitochondria morphological alterations, and the oxidation of pyridine nucleotides and mitochondrial thiols [237]. These effects were completely eliminated by MPTP inhibitors (CsA, DTT, and NEM) and partly eliminated by catalase. The opening of this pore requires the simultaneous oxidation of thiol groups on both sides of the inner mitochondrial membrane [237].
In3+ in experiments with succinate-energized RLM induced mitochondrial swelling, ΔΨmito decline, ROS production, a proton permeability increase, and electron transition inhibition [238]. In3+-induced MPTP was due to the inhibition of the inner membrane proton channels and the stimulation of mitochondrial oxidative stress, resulting in a 15–20% decrease in mitochondrial respiration in states 4, 3, and 3UDNP [238]. The In3+-induced swelling, ΔΨmito decline, and proton permeability increase were markedly eliminated by MPTP inhibitors (ADP and CsA), chelating agents (EGTA and EDTA), and ruthenium red but not by the thiol reagent DTT.
Al(III), Ga(III), and In(III). Cell research. Al(III) exists in water as a hydroxocomplex Al(H2O)63+ that reacts with superoxide radical O2•− to form semireduced radical AlO2(H2O)52+• that depletes mitochondrial Fe followed by the generation of H2O2, O2•−, and OH• [239]. Both Al3+ and aluminum compounds target body tissues resulting in bone mineralization impairment, hemoglobin synthesis impairment, and cardiovascular and neurological disorders [239]. Al3+ ions additionally increased glutamate-induced intracellular Ca2+ overload in rat hippocampal organotypic cultures [240]. Al3+ resulted in an intracellular Ca2+ increase followed by the inhibition of protein kinase B phosphorylation, resulting in cell morphology impairment in PC12 cells [241].
Al3+-induced apoptosis accompanied by the activation of caspases-9, -8, and -3 and oxidative stress manifested as a cell viability decrease, cytochrome c release, and ΔΨmito decline in experiments with rat osteoblasts [242][243]. Al3+ induced an ROS generation increase and mitochondrial damage, resulting in a decrease in SOD and catalase activities and ATP synthesis in hippocampal neuronal cells and human HepG2 cells [244][245][246]. These effects were attenuated by chlorogenic acid [244]. Al3+-induced oxidative stress showed toxic effects that manifested as cell viability collapse, ΔΨmito decline, an increase in ROS production, and a decrease in oxygen consumption and mitochondria enzyme activity in experiments with rat nerve cells and human peripheral blood mononuclear cells [247][248].
Experiments with rats fed with AlCl3 via drinking water showed that Al3+ caused a reduction in the cytoplasm ATP level, a decrease in CI–CIV activity, and disturbance to mitochondrial DNA transcripts, which was followed by the inhibition of the mRNA expression of NADH dehydrogenases 1 and 2, cytochrome b, cytochrome c oxidase subunits 1 and 3, and ATP synthase [249]. By violating the mitochondrial energy metabolism, AlCl3 caused an increase in important liver aspartate and alanine aminotransferases and histopathological lesions. Aluminum nanoparticles (AlNPs), more so than Al3+, induced apoptosis, a cellular viability decrease, a DNA damage increase, mitochondrial dysfunction, and oxidative stress, as well as a decrease in ΔΨmito and reduced glutathione, in experiments with human hepatic HepG2 cells and differentiated HepaRG cells [250].
Ga3+ ions transport into cells via the use of cell Fe3+ transport systems [251]. A decrease in Ga3+-induced cellular iron uptake and intracellular iron homeostasis disruption resulted in mitochondrial function inhibition due to an intracellular ROS production increase and a reduced glutathione decrease as well as iron-containing proteins being targeted in the electronic transport chain [251]. Ga3+ triggers apoptosis by Bax or p53 activation, which was followed by translocation to mitochondria, which results in ΔΨmito loss, an ROS production increase, and cytochrome c release into the intermembrane space and the cytoplasm [251].
Indium(III) compounds were found to induce a cell viability decrease, a lipid peroxidation and ROS production increase, reduced glutathione depletion, an SOD activity decrease, ΔΨmito decline, and oxidative stress damage as well as the expression of apoptotic genes (p53, bax, bcl-2, caspase-3, and caspase-9), oxidative DNA damage, and the formation of condensed chromosomal bodies in experiments in vitro with human lung epithelial A549 cells, lung-derived epithelial (LA-4) cells, human bronchial epithelial BEAS-2B cells, human embryonic kidney 293T cells, and mouse monocyte macrophage RAW 264.7 cells [238][252][253][254][255][256]. In3+ in experiments with human GM5565 skin fibroblasts affected mitochondrial morphology, and organelles became fragmented with a dotted geometry, which may indicate organelle aggregation caused by increased ROS production [257]. Indium oxide nanoparticles were found to have an acute toxic influence on epithelial 16HBE and macrophage (RAW264.7) cells in experiments with rats subjected to particle inhalation [258]. Damage to mitochondria, a rough endoplasmic reticulum, and a cell viability decrease were found.
These trivalent cations (Al3+, Ga3+, and In3+) enter cells using Fe3+ transport systems, which was followed by ROS production with lipid and protein oxidation and iron homeostasis disruption and a reduced glutathione decrease. As a result, damage to iron-containing proteins takes place in the electronic transport chain. There is an entirely different mechanism for inducing both oxidative stress (Figure 1) and apoptosis (Figure 2). The free radical oxidation of thiol groups caused by these metals leads to decreased CI–CIV activity, ATP production, and ΔΨmito decline as well as decreased SOD and catalase activities. The expression of apoptotic genes and oxidative DNA damage accompanies apoptosis induction. Of course, these trivalent metals can be attributed to toxicants disrupting iron and calcium metabolism. This circumstance can harm the hematopoietic system and the nervous system cells. However, accumulating these metals in the body can aggravate diseases associated with the degradation of the nervous and hematopoietic systems and the synthesis of hemoglobin and other iron-containing proteins.
As(III). Mitochondrial research. As(III) (As2O3 or AsO2−) induced a decrease in CI and CII activities, ΔΨmito, and glutathione content, as well as an increase in ROS production, lipid peroxidation, cytochrome c release, and swelling, in isolated RLM or mouse liver mitochondria [259][260][261][262]. Some of these As3+ toxic effects were increased by pyruvate [259] and attenuated by alpha lipoic and ellagic acids [260][263]. AsO2− induced an H2O2 production increase in isolated RHM [264].
As2O3-induced MPTP opening was accompanied by an increase in mitochondrial swelling and Ca2+-induced Ca2+ release, cytochrome c release from the matrix in the inner membrane space, and ΔΨmito decline in succinate-energized RLM [265][266]. However, Ca2+ is required for MPTP opening. CsA, NEM, or RR inhibited MPTP. As3+ did not affect the mitochondrial GSH content in RLM [266]. RLM takes up H2AsO4− via the Pi-dependent pathway and can reduce As5+ to As3+ in participating thioredoxin reductase and reduced glutathione, which is followed by the extrusion of As3+ from mitochondria [267]. This process is more effective with CI substrates than succinate or ADP but is abolished by electron transport inhibitors, uncouplers, ATP synthase inhibitors, and Pi-transport inhibitors.
As(III). Cell research. The available literature analysis makes it possible to attribute As(III) chemical forms (As2O3, AsO2−) to mild thiol reagents. This is the reason for their widespread use in ancient Chinese medicine and in the treatment of certain cancers. Arsenic (As2S2, As2O3, NaAsO2, and Na2HAsO4) and mercury (HgS, HgCl2, and MeHg) are widely used in oriental medicine [268]. It was found that the AsO2−-induced superoxide increase in human myeloid leukemia U937 cells has two contrary pathways: the first leads to the promotion of Nrf2 signaling followed by GSH biosynthesis; the second results in DNA damage and MPTP opening with rapid and massive apoptotic cell death [269][270]. As(III) (AsO2− and As2O3) increased the intracellular Ca2+ concentration and mitochondrial dysfunction due to Ca2+ mobilizing from the ER in human myeloid leukemia U937 cells, human bronchial epithelial HBE cells, and human umbilical and bone marrow mesenchymal stem cells [271][272][273].
As(III) (As2O3 and AsO2−) induced apoptosis with the expression of apoptosis-inducing factors (Bcl-2 and Bcl2), which was followed by a cell viability decrease, the activation of some caspases, and cytochrome c release in the cytoplasm in experiments with L02 hepatocytes, Hepa-1c1c7 cells, Hela cells, mouse insulinoma MIN6 cells, isolated rat hepatocytes, adult rat hepatic stem cells, H9c2 cardiomyocytes, human hepatocellular carcinoma HepG2 cells, colon cancer HT-29 cells, human myeloid leukemia U937 cells, human bronchial epithelial HBE cells, and human umbilical and bone marrow mesenchymal stem cells [155][156][266][271][272][273][274][275][276][277][278][279][280][281][282][283]. However, As(III) (As2O3 and AsO2−) brought about oxidative stress. The stress manifested in MPTP opening, mitochondrial swelling, ΔΨmito decline, a decrease in ATP, reduced glutathione, an ROS production increase, and cytochrome c release in investigations with PC12 cells, Hela cells, isolated rat hepatocytes, rat cardiomyocyte H9c2 cells, L02 hepatocytes, lung bronchial epithelial HBE cells, Hela cells, mouse insulinoma MIN6 cells, laryngeal squamous carcinoma Hep-2 cells, Chang human hepatocytes, mouse embryonic fibroblasts, mouse hepatocytes, human hepatocellular carcinoma HepG2 cells, colon cancer HT-29 cells, cervical cancer cells, primary cultured mouse hepatocytes, and human myeloid leukemia U937 cells [155][156][266][274][276][277][278][279][280][282][283][284][285][286][287][288][289][290][291]. In addition, AsO2− in L02 hepatocytes induced ERK signaling to activate both apoptosis and mitophagy [277] along with ferroptosis in mouse insulinoma MIN6 cells or PC-12 cells [278][292]. Oxygen consumption rates and CI and catalase activity without/with FCCP were increased with low AsO2− but decreased with high AsO2− in experiments with human and mouse primary hepatocytes [288]. Diamide or PAO but not tBHP or AsO2− caused MPTP opening due to the oxidation of ANT critical Cys56 [293].
N-acetyl-L-cysteine, L-ascorbic acid, and selenite (SeO32−) prevented AsO2−-induced apoptosis, oxidative stress, a cell viability decrease, mitochondrial disfunction, cytochrome c release, and an ROS production increase in experiments with L02 hepatocytes, mouse oligodendrocyte precursor cells, human myeloid leukemia U937 cells, and acute promyelocytic leukemia NB4 cells [274][294][295][296]. The effect of SeO32− can be caused by the inhibition of AsO2− transport in NB4 cells [296]. Pterostilbene or tert-butylhydroquinone activating the Nrf2 pathway alleviated similar AsO2−-induced effects in human HaCaT keratinocytes, mouse epidermal JB6 cells, and human epithelial HaCaT cells [297][298]. An increased glutathione biosynthesis capacity prevented As3+-induced apoptosis due to a decrease in caspase activation and cytochrome c release in the cytoplasm in mouse liver hepatoma Hepa-1c1c7 cells [275]. Autophagy can inhibit As2O3-induced apoptosis in HL60 cells in the initiation stage [299]. Metallothionein or methyl donors (betaine, methionine, and folic acid) alleviated the As3+-induced ΔΨmito decline and ROS production increase in PC12 cells or isolated rat hepatocytes [284][285]. The ferroptosis inhibitor Fer-1 attenuated the similar toxic effects of As2O3 on rat cardiomyocyte H9c2 cells [286]. Experiments with rats co-exposed to NaAsO2 and NaF showed oxidative stress and autophagy in myocardial tissue and cells [282]. As3+-induced oxidative stress and the apoptosis mitochondrial pathway were alleviated in experiments with the glutathione treatment of chronic arsenic-exposed mice [300].
Arsenic nanoparticles induced by apoptosis, mitochondrial swelling, ΔΨmito decline, a glutathione decrease, an ROS production increase, and membrane integrity were disturbed in isolated rat hepatocytes [301].
Sb(III). Mitochondrial and cell research. Experiments with Sb3+-treated mitochondria isolated from A549 cells showed an increase in SOD activity and a decrease in CI and CIII activities, glutathione peroxidase, glutathione reductase, and thioredoxin reductase [302]. The compounds and complexes of pentavalent and trivalent antimony with potassium antimonyl tartrate are used to treat leishmaniasis caused by protozoan parasites [303][304][305]. Sb(III) (Sb2O3 and SbCl3) induced apoptosis and cell viability loss; an increase in ROS production; and a decrease in the cytoplasm glutathione level, ΔΨmito, and ATP content in human lung adenocarcinoma A549 cells, human embryonic kidney HEK-293 cells, and CCRF-CEM line cells [302][306][307]. Potassium antimonyl tartrate (K2[SbC2H2(O)2(COO)2]2) and As2O3 had similar effects in experiments with human lymphoid tumoral cells and human myeloid leukemic HL60 cells [308][309]. Sb3+-induced apoptosis in CCRF-CEM cells increased in the presence of both buthionine sulfoximine (a gamma-glutamylcysteine synthetase inhibitor) and sodium ascorbate, which reduce intracellular glutathione levels in human myeloid leukemic HL60 cells [307][309]. The metal-induced ROS production increase and cell viability decrease showed the order of As3+ > As5+ > Sb3+ > Sb5+, and these effects were the main cause of Sb cytotoxicity in these cells [306].
As(III), similar to the other heavy metals above, induced oxidative stress (Figure 1) accompanied by a decrease in CI and CII activities, MPTP opening, an ROS production increase, and an ATP production and ΔΨmito decline. However, As3+ (unlike the heavy metals discussed above) can be classified as a mild thiol effector due to its weak influence on mitochondrial GSH content. As(III) similar to Pb2+ increases cytoplasm [Ca2+] mobilizing Ca2+ from the ER, which is followed by mitochondrial dysfunction due to the calcium overload of those organelles. That elimination of the As(III)-induced oxidative stress and apoptosis consequences in the presence of NAC, ascorbate, and selenite is probably due to the significant reversibility of these As(III)-induced processes. It should be noted that this circumstance is indicated by preventing As3+-induced apoptosis due to the acceleration of the processes of glutathione biosynthesis or the action of metallothioneins or methyl donors on cells. A few studies on Sb(III) compounds found effects similar to those of As(III). Undoubtedly, the moderate binding of As(III) to the thiol groups of various proteins and enzyme complexes has become the reason for the recent rapid growth in research on the search for and synthesis of arsenic-containing drugs that selectively suppress the growth of cancer cells for the treatment of oncological diseases.
Cr(VI). Mitochondrial research. Cr2O72− induced a state 3 and 3UFCCP respiration decrease, an ROS production increase, and ΔΨmito decline in isolated RLM energized by CI and CII substrates [310][311]. However, state 4 respiration increased and peaked at 250 μM K2Cr2O7, which was followed by a sharp decrease in respiration [311]. Dichromate weakly affected the inner membrane passive proton permeability; however, the FCCP-induced permeability of the IMM was partially reduced by Cr2O72− [311]. RLM took up Cr(VI), which reduced further to Cr(V) in the matrix [312]. Glutathione eliminated these effects of Cr2O72− [76][310]. Cr2O72− induced the state 3 inhibition and ΔΨmito decline in mitochondria isolated from L02 hepatocytes [313].
Cr(VI). Cell research. Cr2O72− caused caspase-3 dependent apoptosis, and mitochondrial dysfunction was accompanied by a cell viability decrease, MPTP opening, mitophagy, a ΔΨmito decline, energy metabolism disturbance with an ATP production decrease, cytochrome c release, a CI–CIII activity decrease, and an intracellular Ca2+ increase due to a mitochondrial ROS production increase and endoplasmic reticulum (ER) stress, followed by Ca2+ release, in experiments with L02 hepatocytes [313][314][315][316][317][318][319][320][321][322]. Cr(VI) (CrO42− and Cr2O72−) induced mitochondrial apoptosis, and this was dependent on both caspase-3 with a preceding p53 signal and mitophagy. Oxidative stress, Ca2+ overload (due to mitochondrial and ER stress), MPTP opening, a cell viability decrease, CI and CII inhibition, a mitochondrial ATP production decrease, ΔΨmito decline, a mitochondrial swelling increase, cytochrome c release, and an ROS production and lipid peroxidation increase were also observed here in experiments with mouse SSCs/progenitors, human lung A549 carcinoma epithelial cells, human lung H1299 adenocarcinoma cells, human neuroblastoma SH-SY5Y cells, human bronchial epithelial BEAS-2B cells, cultured cerebellar granule neurons, human hepatoma Hep3B cells, rat granulosa cells, mouse epidermal Cl 41 cells, human bronchoalveolar carcinoma H358 cells, and human myeloid leukemia U937 cells [316][323][324][325][326][327][328][329][330][331][332][333][334]. A Cr2O72− induced decrease in Ca2+, Mg2+-ATPase and Na+, K+-ATPase activities was found in human hepatoma Hep3B cells [316]. Some of these Cr2O72−-induced effects were inhibited by N-acetyl-L cysteine, vitamin C, BAPTA-AM, and clusterin (disulfide-linked multifunctional glycoprotein). Zn2+ attenuated the CrO42−-induced mitochondrial caspase-3 apoptosis and oxidative stress that manifested as ΔΨmito decline and an ROS or H2O2 production increase in human tumor Hep-2cells [335].
Cr(VI) compounds are of industrial origin and very rarely occur in nature in the form of crocoite mineralade, PbCrO4. Being the most potent oxidizing agent, Cr2O72− induced oxidative stress (Figure 1) in addition to lysosomal membrane rupture, GSH oxidation, CI and CII inhibition, a decline in ΔΨmito and ATP production, MPTP opening, ROS production, and an increase in lipid peroxidation in experiments with various cells in vitro. At the same time, under acting Cr(VI) compounds, as in the case of the heavy metals considered above, apoptosis (Figure 2) was induced due to caspase-3 activation with a preceding p53 signal and mitophagy. Antioxidants and ROS scavengers prevented these cytotoxic effects. Cr(VI) toxicity may increase due to the activation of nitrosylation processes in cells, which was followed by the activation of apoptosis by a mechanism involving S-nitrosylation and nitric oxide-dependent stabilization of the Bcl-2 protein [336]. Therefore, cell damage caused by Cr(VI) compounds can provoke carcinogenesis, leading to oncological diseases, including lung cancer, due to the malignant transformation of human lung epithelial cells.
U(VI). Mitochondrial and cell research. UO22+ attenuated mitochondrial respiration in states 3 and 3UDNP and induced both a H2O2 formation increase and a slight calcium retention increase in succinate-energized RLM [337]. UO22+ induced an ROS production and lipid peroxidation increase as well as a reduced glutathione content and ΔΨmito decrease in the isolated kidney mitochondria of rats injected with uranyl acetate [338]. The toxicity of UO22+ manifested as CII and CIII inhibition, mitochondrial swelling, ΔΨmito decline, and an increase in H2O2 and ROS production and lipid peroxidation as well as a decrease in cell viability, reduced glutathione content, mitochondrial ATP content, the ATP/ADP ratio, cytochrome c release, and outer mitochondrial membrane damage in experiments with primary rat hepatocytes, human dermal fibroblast primary cells, and isolated rat kidney mitochondria (RKM) energized by CI and CII substrates [338][339][340][341]. MPTP inhibitors (carnitine, CsA, and trifluoperazine), antioxidants (catalase), ROS scavengers (mannitol and DMSO), or a U6+ reduction inhibitor (Ca2+) prevented the UO22+-induced ΔΨmito decrease and lysosomal membrane hepatocyte damage [339]. Ca2+ blocked the glutathione or cysteine capacity to reduce U5+ and U6+ to U4+ to form O2•− in these hepatocytes [339]. Beta-glucan and butylated hydroxyl toluene prevented both these toxic UO22+ effects and the UO22+-induced outer mitochondrial membrane damage in RKM [342].
Uranyl binding with cyt b5, cyt c, and the cyt b5–cyt c complex may indicate that UO22+-induced apoptosis forms a dynamic cyt b5–cyt c complex [343]. UO22+ induced apoptosis with the activation of caspases-3, -8, -9, and -10 to indicate a mitochondria-dependent signaling pathway in rat kidney NRK-52E proximal cells, rat hepatic BRL cells, HEp-2 cells, and human dermal fibroblast primary cells [337][341][344][345]. There was also UO22+-induced oxidative stress accompanied by a cell viability decrease, an ROS production increase, a lipid peroxidation increase, a reduced glutathione content decrease, and ΔΨmito collapse. Zn2+ (an effective heavy metal poisoning antidote) protected against UO22+-induced apoptosis in human kidney HK-2 cells [346]. In addition, Zn2+ prevented the UO22+-induced cell viability decrease, cytochrome c release to the cytoplasm, ΔΨmito decline, lactate dehydrogenase (LDH) depletion, and increase in ROS production and catalase and glutathione concentrations [346].
UO22+ (similar to heavy metals) initiated apoptotic processes with the activation of caspases-3 and -9 and formed a dynamic cyt b5–cyt c complex. Moreover, uranyl-induced mitochondrial oxidative stress (Figure 1) manifested as an ROS production and lipid peroxidation increase, a GSH decrease, CII and CIII inhibition, MPTP opening, and ΔΨmito decline. The cytotoxic effects of UO22+ were markedly attenuated in experiments with MPTP inhibitors, antioxidants, and ROS scavengers. High uranyl toxicity was found for some bacteria [347]. In addition, uranyl disrupts cellular calcium metabolism. One can hypothesize that uranyl toxicity (together with natural radioactivity) may manifest itself through disturbances in calcium-dependent metabolic processes, exacerbating metabolic disorders. The toxicity of UO2 was higher than the toxicity of Al and comparable to the toxicity of Cu, Zn, and Pb, but it was lower than the toxicity of Cd and Ag [348].
Modified heavy metals. Cell research. Reduced glutathione was shown to react directly with Ag+ ions [89]. Tl3+ ions unlike Tl+ ones oxidized glutathione [349]. The toxic effects of MeHg were more potent than those of Hg2+ on zebrafish ZF4 and human gingival fibroblasts [147][157]. The co-exposure of AgNPs, Cd2+, and Hg2+ showed increased toxicity in experiments with human hepatocarcinoma HepG2 cells, with AgNP + Hg2+ being less toxic than AgNPs + Cd2+ [350]. Nanoparticles (CdO or Ag) inhibited succinate dehydrogenase in BRL-3A hepatocytes [99]. Nano-carbon black with Pb2+ particles simulating the atmosphere fine particles formed a special complex that activated apoptotic signaling pathways, impaired ΔΨmito, and inhibited the lysosomal function in rat alveolar macrophages [351]. Pb4+ as Pb(acetate)4 increased intracellular human neuroglobin and ROS production in breast cancer MCF-7 cells and induced mitochondrial apoptosis [352]. Trialkyllead compounds similar to Ca2+ with valinomycin induced an increase in mitochondrial swelling, state 4 respiration, and K+ efflux, as well as a decrease in state 3 respiration, in succinate-energized RLM injected in KCl but not in a KNO3 medium [353][354][355]. Minerals containing heavy metals with a maximum oxidation state (Tl3+ and Pb4+) do not occur in nature, and their use in research is purely for laboratory use. Ph3Sb(V)O in complex with carvacrol was found to reduce in participating glutathione to a toxic form containing a Ph3Sb(III) complex that resulted in apoptosis, mitochondrion damage, and mitochondrial membrane permeabilization in human breast adenocarcinoma MCF-7 cells [356]. The AlF4− complex being isomorphous to Pi inhibited beef heart mitochondrial F1-ATPase, pig kidney Na+, K+-ATPase, plasmalemmal stomach smooth muscle Ca2+, and Mg2+-ATPase [357][358][359]. Gold(I)-derived complexes synthesized on the basis of Au[P(Ph)3]+Cl− were proven to be effective trypanothione reductase inhibitors and proposed to treat leishmaniasis [360]. These gold complexes induced oxidative stress with an ROS production increase, mitochondrial damage, and a mitochondrial respiration decrease in experiments with human monocyte-derived macrophage THP-1 cells infected by leishmaniasis parasites.
This entry is adapted from the peer-reviewed paper 10.3390/ijms241914459
References
- Riley, M.V.; Lehninger, A.L. Changes in sulfhydryl groups of rat liver mitochondria during swelling and contraction. J. Biol. Chem. 1964, 239, 2083–2089.
- Faddeeva, M.D.; Beliaeva, T.N.; Sokolovskaia, E.L. Effect of a number of biologically active substances on the activity of membrane-bound Na+,K+-ATPase from the bovine brain. Tsitologiia 1987, 29, 576–581. (In Russian)
- Higashida, H.; Semba, R.K.; Niwa, F.; Kashiwamata, S. Mitochondrial malate dehydrogenase of bovine cerebrum. Characterization and mechanisms of inhibition by silver ions. J. Biochem. 1975, 78, 989–999.
- Kone, B.C.; Kaleta, M.; Gullans, S.R. Silver ion Ag+-induced increases in cell membrane K+ and Na+ permeability in the renal proximal tubule: Reversal by thiol reagents. J. Membr. Biol. 1988, 102, 11–19.
- Kone, B.C.; Brenner, R.M.; Gullans, S.R. Sulfhydryl-reactive heavy metals increase cell membrane K+ and Ca2+ transport in renal proximal tubule. J. Membr. Biol. 1990, 113, 1–12.
- Williams, R.J.V. The biochemistry of sodium, potassium, magnesium, and calcium. Q. Rev. Chem. Soc. 1970, 3, 331–365.
- Douglas, K.T.; Bunni, M.A.; Baindur, S.R. Thallium in biochemistry. Int. J. Biochem. 1990, 22, 429–438.
- Skul’skiĭ, I.A. The isomorphism of univalent thallium and potassium in membrane transport processes. Tsitologiia 1991, 33, 118–129. (In Russian)
- Korotkov, S.M. Mitochondria as a Key Intracellular Target of Thallium Toxicity; Academic Press: Cambridge, MA, USA, 2022; pp. 1–261. Available online: https://www.elsevier.com/books/mitochondria-as-a-key-intracellular-target-of-thallium-toxicity/korotkov/978-0-323-95531-7 (accessed on 1 June 2022).
- Mulkey, J.P.; Oehme, F.W. A review of thallium toxicity. Vet. Hum. Toxicol. 1993, 35, 445–453.
- Zhang, D.; Gopalakrishnan, S.M.; Freiberg, G.; Surowy, C.S. A thallium transport FLIPR-based assay for the identification of KCC2-positive modulators. J. Biomol. Screen. 2010, 15, 177–184.
- Blain, R.; Kazantzis, G. Thallium. In Handbook on the Toxicology of Metals—Volume II: Specific Metals; Nordberg, G.F., Fowler, B.A.L., Nordberg, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 1229–1240.
- Nriagu, J.O. Thallium in the Environment; Wiley Series in Advances in Environmental Science and Technology; Wiley: New York, NY, USA, 1998; pp. 1–284.
- McCall, D.; Zimmer, L.J.; Katz, A.M. Kinetics of thallium exchange in cultured rat myocardial cells. Circ. Res. 1985, 56, 370–376.
- Herman, M.M.; Bensch, K.G. Light and electron microscopic studies of acute and chronic thallium intoxication in rats. Toxicol. Appl. Pharmacol. 1967, 10, 199–222.
- Mourelle, M.; Favari, L.; Amezcua, J.L. Protection against thallium hepatotoxicity by silymarin. J. Appl. Toxicol. 1988, 8, 351–354.
- Leung, K.M.; Ooi, V.E. Studies on thallium toxicity, its tissue distribution and histopathological effects in rats. Chemosphere 2000, 41, 155–159.
- Li, S.; Huang, W.; Duan, Y.; Xing, J.; Zhou, Y. Human fatality due to thallium poisoning: Autopsy, microscopy, and mass spectrometry assays. J. Forensic Sci. 2015, 60, 247–251.
- Repetto, G.; Sanz, P.; Repetto, M. Comparative in vitro effects of sodium arsenite and sodium arsenate on neuroblastoma cells. Toxicology 1994, 92, 143–153.
- Puga Molina, L.C.; Verstraeten, S.V. Thallium(III)-mediated changes in membrane physical properties and lipid oxidation affect cardiolipin-cytochrome c interactions. Biochim. Biophys. Acta 2008, 1778, 2157–2164.
- Rodríguez-Mercado, J.J.; Altamirano-Lozano, M.A. Genetic toxicology of thallium: A review. Drug Chem. Toxicol. 2013, 36, 369–383.
- Zierold, K. Heavy metal cytotoxicity studied by electron probe X-ray microanalysis of cultured rat hepatocytes. Toxicol. In Vitro 2000, 14, 557–563.
- Korotkov, S.M.; Nesterov, V.P.; Belostotskaya, G.B.; Brailovskaya, I.V.; Novozhilov, A.V.; Sobol, C.V. Influence of Tl+ on the movement of Ca2+ and Na+ ions across membranes of rat neonatal cardiomyocytes and rat heart mitochondria. Ukr. Biochem. J. 2020, 92, 41–55.
- Melnick, R.L.; Monti, L.G.; Motzkin, S.M. Uncoupling of mitochondrial oxidative phosphorylation by thallium. Biochem. Biophys. Res. Commun. 1976, 69, 68–73.
- Woods, J.S.; Fowler, B.A. Alteration of hepatocellular structure and function by thallium chloride: Ultrastructural, morphometric, and biochemical studies. Toxicol. Appl. Pharmacol. 1986, 83, 218–229.
- Korotkov, S.M.; Brailovskaya, I.V.; Kormilitsyn, B.N.; Furaev, V.V. Tl+ showed negligible interaction with inner membrane sulfhydryl groups of rat liver mitochondria, but formed complexes with matrix proteins. J. Biochem. Mol. Toxicol. 2014, 28, 149–156.
- Perrin, D.D. Stability Constants of Metal-Ion Complexes—Part B—Organic Ligands; Pergamon: Oxford, UK, 1979; pp. 1–1263.
- Weinberg, J.M.; Harding, P.G.; Humes, H.D. Mitochondrial bioenergetics during the initiation of mercuric chloride-induced renal injury. I. Direct effects of in vitro mercuric chloride on renal mitochondrial function. J. Biol. Chem. 1982, 257, 60–67.
- Chávez, E.; Holguín, J.A. Mitochondrial calcium release as induced by Hg2+. J. Biol. Chem. 1988, 263, 3582–3587.
- Lund, B.O.; Miller, D.M.; Woods, J.S. Studies on Hg(II)-induced H2O2 formation and oxidative stress in vivo and in vitro in rat kidney mitochondria. Biochem. Pharmacol. 1993, 45, 2017–2024.
- Chávez, R.; Corona, N.; García, C.; Chávez, E. The effect of cyclosporin A on Hg2+-poisoning mitochondria. In vivo and in vitro studies. Comp. Biochem. Physiol. C Pharmacol. Toxicol. Endocrinol. 1994, 107, 429–434.
- Southard, J.H.; Penniston, J.T.; Green, D.E. Induction of transmembrane proton transfer by mercurials in mitochondria. I. Ion movements accompanying transmembrane proton transfer. J. Biol. Chem. 1973, 248, 3546–3550.
- Reyes-Vivas, H.; Lopez-Moreno, F.; Chávez, E. Protective effect of diethyldithiocarbamate on mercury-induced toxicity in kidney mitochondria. Comp. Biochem. Physiol. C Pharmacol. Toxicol. Endocrinol. 1996, 113, 349–352.
- Shenker, B.J.; Guo, T.L.; Shapiro, I.M. Low-level methylmercury exposure causes human T-cells to undergo apoptosis: Evidence of mitochondrial dysfunction. Environ. Res. 1998, 77, 149–159.
- Chandler, J.E.; Messer, H.H.; Ellender, G. Cytotoxicity of gallium and indium ions compared with mercuric ion. J. Dent. Res. 1994, 73, 1554–1559.
- Woods, J.S.; Ellis, M.E. Up-regulation of glutathione synthesis in rat kidney by methyl mercury. Relationship to mercury-induced oxidative stress. Biochem. Pharmacol. 1995, 50, 1719–1724.
- Korotkov, S.M.; Skulskii, I.A.; Glazunov, V.V. Cd2+ effects on respiration and swelling of rat liver mitochondria were modified by monovalent cations. J. Inorg. Biochem. 1998, 70, 17–23.
- Korotkov, S.M. The Effect of Cadmium on Oxidative Phosphorylation and Ion Transport in Mitochondria. Ph.D. Thesis, Russian Federation, St. Petersburg, Russia, 1987; pp. 1–24. (In Russian).
- Lee, W.K.; Spielmann, M.; Bork, U.; Thévenod, F. Cd2+-induced swelling-contraction dynamics in isolated kidney cortex mitochondria: Role of Ca2+ uniporter, K+ cycling, and protonmotive force. Am. J. Physiol. Cell Physiol. 2005, 289, C656–C664.
- Al-Nasser, I.A. Cadmium hepatotoxicity and alterations of the mitochondrial function. J. Toxicol. Clin. Toxicol. 2000, 38, 407–413.
- Rasheed, B.K.; Diwan, J.J.; Sanadi, D.R. Activation of potassium ion transport in mitochondria by cadmium ion. Eur. J. Biochem. 1984, 144, 643–647.
- Belyaeva, E.A.; Korotkov, S.M.; Saris, N.E. In vitro modulation of heavy metal-induced rat liver mitochondria dysfunction: A comparison of copper and mercury with cadmium. J. Trace Elem. Med. Biol. 2011, 25, S63–S73.
- Skul’skiĭ, I.A.; Korotkov, S.M.; Glazunov, V.V.; Ivanova, T.I.; Savina, M.V. Effect of cadmium ions on the respiration, cation transport and morphology of the liver mitochondria in the rat and the lamprey. Tsitologiia 1988, 30, 956–962. (In Russian)
- Belyaeva, E.A.; Glazunov, V.V.; Korotkov, S.M. Cyclosporin A-sensitive permeability transition pore is involved in Cd2+-induced dysfunction of isolated rat liver mitochondria: Doubts no more. Arch. Biochem. Biophys. 2002, 405, 252–264.
- Koike, H.; Shinohara, Y.; Terada, H. Why is inorganic phosphate necessary for uncoupling of oxidative phosphorylation by Cd2+ in rat liver mitochondria? Biochim. Biophys. Acta 1991, 1060, 75–81.
- Shemarova, I.V.; Korotkov, S.M.; Nesterov, V.P. Effect of oxidative processes in mitochondria on contractility of heart muscle of the frog Rana temporaria. Actions of Cd2+. J. Evol. Biochem. Physiol. 2011, 47, 360–365.
- Korotkov, S.M.; Nesterov, V.P.; Emel’yanova, L.V.; Ryabchikov, N.N. Involvement of SH-groups during interaction of diazoxide with the inner membrane of rat heart mitochondria. Dokl. Biochem. Biophys. 2007, 415, 206–210.
- Dorta, D.J.; Leite, S.; DeMarco, K.C.; Prado, I.M.; Rodrigues, T.; Mingatto, F.E.; Uyemura, S.A.; Santos, A.C.; Curti, C. A proposed sequence of events for cadmium-induced mitochondrial impairment. J. Inorg. Biochem. 2003, 97, 251–257.
- Miccadei, S.; Floridi, A. Sites of inhibition of mitochondrial electron transport by cadmium. Chem. Biol. Interact. 1993, 89, 159–167.
- Bagchi, D.; Vuchetich, P.J.; Bagchi, M.; Hassoun, E.A.; Tran, M.X.; Tang, L.; Stohs, S.J. Induction of oxidative stress by chronic administration of sodium dichromate and cadmium chloride to rats. Free Radic. Biol. Med. 1997, 22, 471–478.
- Jay, D.; Zamorano, R.; Muñoz, E.; Gleason, R.; Boldu, J.L. Study of the interaction of cadmium with membrane-bound succinate dehydrogenase. J. Bioenerg. Biomembr. 1991, 23, 381–389.
- Viarengo, A.; Nicotera, P. Possible role of Ca2+ in heavy metal cytotoxicity. Comp. Biochem. Physiol. C Comp. Pharmacol. Toxicol. 1991, 100, 81–84.
- He, L.; Poblenz, A.T.; Medrano, C.J.; Fox, D.A. Lead and calcium produce rod photoreceptor cell apoptosis by opening the mitochondrial permeability transition pore. J. Biol. Chem. 2000, 275, 12175–12184.
- Fox, D.A.; He, L.; Poblenz, A.T.; Medrano, C.J.; Blocker, Y.S.; Srivastava, D. Lead-induced alterations in retinal cGMP phosphodiesterase trigger calcium overload, mitochondrial dysfunction and rod photoreceptor apoptosis. Toxicol. Lett. 1998, 102–103, 359–361.
- Kapoor, S.C.; van Rossum, G.D.; O’Neill, K.J.; Mercorella, I. Uptake of inorganic lead in vitro by isolated mitochondria and tissue slices of rat renal cortex. Biochem. Pharmacol. 1985, 34, 1439–1448.
- Chávez, E.; Jay, D.; Bravo, C. The mechanism of lead-induced mitochondrial Ca2+ efflux. J. Bioenerg. Biomembr. 1987, 19, 285–295.
- van Rossum, G.D.; Kapoor, S.C.; Rabinowitz, M.S. Effects of inorganic lead in vitro on ion exchanges and respiratory metabolism of rat kidney cortex. Arch. Toxicol. 1985, 56, 175–181.
- Kapoor, S.C.; van Rossum, G.D. Effects of Pb2+ added in vitro on Ca2+ movements in isolated mitochondria and slices of rat kidney cortex. Biochem. Pharmacol. 1984, 33, 1771–1778.
- Scott, K.M.; Hwang, K.M.; Jurkowitz, M.; Brierley, G.P. Ion transport by heart mitochondria. 23. The effects of lead on mitochondrial reactions. Arch. Biochem. Biophys. 1971, 147, 557–567.
- Cardona, E.; Lessler, M.A.; Brierley, G.P. Mitochondrial oxidative phosphorylation: Interaction of lead and inorganic phosphate. Proc. Soc. Exp. Biol. Med. 1971, 136, 300–304.
- Castaño, R.; Culebras, C.; Zazueta, C.; Chávez, E. Protective role of chlorpromazine on lead-induced damage to heart mitochondria. Comp. Biochem. Physiol. C Comp. Pharmacol. Toxicol. 1991, 99, 379–381.
- Sabbioni, E.; Marafante, E. Identification of lead-binding components in rat liver: In vivo study. Chem. Biol. Interact. 1976, 15, 1–20.
- Dini, L.; Giudetti, A.M.; Ruzittu, M.; Gnoni, G.V.; Zara, V. Citrate carrier and lipogenic enzyme activities in lead nitrate-induced proliferative and apoptotic phase in rat liver. Biochem. Mol. Biol. Int. 1999, 47, 607–614.
- Simons, T.J.B. Lead-calcium interactions and lead toxicity. In Calcium in Drug Actions; Baker, P.F., Ed.; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 1988; Volume 83, pp. 509–525.
- Simons, T.J. Lead-calcium interactions in cellular lead toxicity. Neurotoxicology 1993, 14, 77–85.
- Miyahara, M.; Utsumi, K. Oxidative phosphorylation controlled by potassium in rat liver mitochondria. Cell Struct. Funct. 1975, 1, 51–59.
- Dominguez, M.C.; Sole, E.; Goñi, C.; Ballabriga, A. Effect of aluminum and lead salts on lipid peroxidation and cell survival in human skin fibroblasts. Biol. Trace Elem. Res. 1995, 47, 57–67.
- Verstraeten, S.V.; Golub, M.S.; Keen, C.L.; Oteiza, P.I. Myelin is a preferential target of aluminum-mediated oxidative damage. Arch. Biochem. Biophys. 1997, 344, 289–294.
- Leikin, Y.N.; Zharova, T.V.; Tjulina, O.V. Novel oxaloacetate effect on mitochondrial Ca2+ movement. FEBS Lett. 1993, 331, 35–37.
- Novgorodov, S.A.; Kultayeva, E.V.; Yaguzhinsky, L.S.; Lemeshko, V.V. Ion permeability induction by the SH cross-linking reagents in rat liver mitochondria is inhibited by the free radical scavenger, butylhydroxytoluene. J. Bioenerg. Biomembr. 1987, 19, 191–202.
- Csillag, A. The effect of arsenite on rat-liver mitochondria. FEBS Lett. 1971, 17, 342–344.
- Guerrieri, F.; Papa, S. Effect of thiol reagents on the proton conductivity of the H+-ATPase of mitochondria. Eur. J. Biochem. 1982, 128, 9–13.
- Rein, K.A.; Borrebaek, B.; Bremer, J. Arsenite inhibits beta-oxidation in isolated rat liver mitochondria. Biochim. Biophys. Acta 1979, 574, 487–494.
- Costantini, P.; Chernyak, B.V.; Petronilli, V.; Bernardi, P. Modulation of the mitochondrial permeability transition pore by pyridine nucleotides and dithiol oxidation at two separate sites. J. Biol. Chem. 1996, 271, 6746–6751.
- Kowaltowski, A.J.; Castilho, R.F.; Grijalba, M.T.; Bechara, E.J.; Vercesi, A.E. Effect of inorganic phosphate concentration on the nature of inner mitochondrial membrane alterations mediated by Ca2+ ions. A proposed model for phosphate-stimulated lipid peroxidation. J. Biol. Chem. 1996, 271, 2929–2934.
- Pourahmad, J.; O’Brien, P.J. Biological reactive intermediates that mediate chromium (VI) toxicity. Adv. Exp. Med. Biol. 2001, 500, 203–207.
- Susa, N.; Ueno, S.; Furukawa, Y. Protective effect of diethyldithiocarbamate pretreatment on chromium (VI)-induced cytotoxicity and lipid peroxidation in primary cultures of rat hepatocytes. J. Vet. Med. Sci. 1998, 60, 71–76.
- Tsunoda, K.; Kozuka, S.; Kawai, K. Mitochondria toxicity of silver ion as the molecular mechanism for the antifungal activity. Dep. Bull. Pap. 2017, 53, 87–97. Available online: https://core.ac.uk/display/236360138? (accessed on 1 June 2022).
- Yuan, L.; Gao, T.; He, H.; Jiang, F.L.; Liu, Y. Silver ion-induced mitochondrial dysfunction via a nonspecific pathway. Toxicol. Res. 2017, 6, 621–630.
- Almofti, M.R.; Ichikawa, T.; Yamashita, K.; Terada, H.; Shinohara, Y. Silver ion induces a cyclosporine a-insensitive permeability transition in rat liver mitochondria and release of apoptogenic cytochrome C. J. Biochem. 2003, 134, 43–49.
- Kawashima, S.; Yamamoto, T.; Horiuchi, Y.; Fujiwara, K.; Gouda, S.; Yoshimura, Y.; Yamamoto, A.; Inotani, Y.; Yamashita, K.; Kitamura, S.; et al. S-15176 and its methylated derivative suppress the CsA-insensitive mitochondrial permeability transition and subsequent cytochrome c release induced by silver ion, and show weak protonophoric activity. Mol. Cell. Biochem. 2011, 358, 45–51.
- Dong, P.; Li, J.H.; Xu, S.P.; Wu, X.J.; Xiang, X.; Yang, Q.Q.; Jin, J.C.; Liu, Y.; Jiang, F.L. Mitochondrial dysfunction induced by ultra-small silver nanoclusters with a distinct toxic mechanism. J. Hazard. Mater. 2016, 308, 139–148.
- Teodoro, J.S.; Simões, A.M.; Duarte, F.V.; Rolo, A.P.; Murdoch, R.C.; Hussain, S.M.; Palmeira, C.M. Assessment of the toxicity of silver nanoparticles in vitro: A mitochondrial perspective. Toxicol. In Vitro 2011, 25, 664–670.
- Wang, F.; Chen, Z.; Wang, Y.; Ma, C.; Bi, L.; Song, M.; Jiang, G. Silver Nanoparticles Induce Apoptosis in HepG2 Cells through Particle-Specific Effects on Mitochondria. Environ. Sci. Technol. 2022, 56, 5706–5713.
- da Silva Ferreira, V.; Eugenio, M.F.C.; Del Nery Dos Santos, E.; de Souza, W.; Sant’Anna, C. Cellular toxicology and mechanism of the response to silver-based nanoparticle exposure in Ewing’s sarcoma cells. Nanotechnology 2021, 32, 115101.
- Sidambaram, P.; Colleran, J. Evaluating the anticancer properties and real-time electrochemical extracellular bio-speciation of bis (1,10-phenanthroline) silver (I) acetate monohydrate in the presence of A549 lung cancer cells. Biosens. Bioelectron. 2021, 175, 112876.
- Michaeloudes, C.; Seiffert, J.; Chen, S.; Ruenraroengsak, P.; Bey, L.; Theodorou, I.G.; Ryan, M.; Cui, X.; Zhang, J.; Shaffer, M.; et al. Effect of silver nanospheres and nanowires on human airway smooth muscle cells: Role of sulfidation. Nanoscale Adv. 2020, 2, 5635–5647.
- Buttacavoli, M.; Albanese, N.N.; Di Cara, G.; Alduina, R.; Faleri, C.; Gallo, M.; Pizzolanti, G.; Gallo, G.; Feo, S.; Baldi, F.; et al. Anticancer activity of biogenerated silver nanoparticles: An integrated proteomic investigation. Oncotarget 2017, 9, 9685–9705.
- Ahlberg, S.; Rancan, F.; Epple, M.; Loza, K.; Höppe, D.; Lademann, J.; Vogt, A.; Kleuser, B.; Gerecke, C.; Meinke, M.C. Comparison of different methods to study effects of silver nanoparticles on the pro- and antioxidant status of human keratinocytes and fibroblasts. Methods 2016, 109, 55–63.
- Jiang, X.; Foldbjerg, R.; Miclaus, T.; Wang, L.; Singh, R.; Hayashi, Y.; Sutherland, D.; Chen, C.; Autrup, H.; Beer, C. Multi-platform genotoxicity analysis of silver nanoparticles in the model cell line CHO-K1. Toxicol. Lett. 2013, 222, 55–63.
- Barbasz, A.; Oćwieja, M.; Roman, M. Toxicity of silver nanoparticles towards tumoral human cell lines U-937 and HL-60. Colloids Surf. B Biointerfaces 2017, 156, 397–404.
- Kaplan, A.; Akalin Ciftci, G.; Kutlu, H.M. The apoptotic and genomic studies on A549 cell line induced by silver nitrate. Tumour Biol. 2017, 39, 1–12.
- Piao, M.J.; Kang, K.A.; Lee, I.K.; Kim, H.S.; Kim, S.; Choi, J.Y.; Choi, J.; Hyun, J.W. Silver nanoparticles induce oxidative cell damage in human liver cells through inhibition of reduced glutathione and induction of mitochondria-involved apoptosis. Toxicol. Lett. 2011, 201, 92–100.
- Inoue, T.; Suzuki, Y.; Yoshimaru, T.; Ra, C. Ca2+-dependent mast cell death induced by Ag(I) via cardiolipin oxidation and ATP depletion. J. Leukoc. Biol. 2009, 86, 167–179.
- Miyayama, T.; Arai, Y.; Suzuki, N.; Hirano, S. Mitochondrial electron transport is inhibited by disappearance of metallothionein in human bronchial epithelial cells following exposure to silver nitrate. Toxicology 2013, 305, 20–29.
- Cortese-Krott, M.M.; Münchow, M.; Pirev, E.; Hessner, F.; Bozkurt, A.; Uciechowski, P.; Pallua, N.; Kröncke, K.D.; Suschek, C.V. Silver ions induce oxidative stress and intracellular zinc release in human skin fibroblasts. Free Radic. Biol. Med. 2009, 47, 1570–1577.
- Wang, L.; Mello, D.F.; Zucker, R.M.; Rivera, N.A.; Rogers, N.M.K.; Geitner, N.K.; Boyes, W.K.; Wiesner, M.R.; Hsu-Kim, H.; Meyer, J.N. Lack of detectable direct effects of silver and silver nanoparticles on mitochondria in mouse hepatocytes. Environ. Sci. Technol. 2021, 55, 11166–11175.
- Li, Y.; Wang, W.X. Uptake, intracellular dissolution, and cytotoxicity of silver nanowires in cell models. Chemosphere 2021, 281, 130762.
- Hussain, S.M.; Hess, K.L.; Gearhart, J.M.; Geiss, K.T.; Schlager, J.J. In vitro toxicity of nanoparticles in BRL 3A rat liver cells. Toxicol. In Vitro 2005, 19, 975–983.
- Das, B.; Tripathy, S.; Adhikary, J.; Chattopadhyay, S.; Mandal, D.; Dash, S.K.; Das, S.; Dey, A.; Dey, S.K.; Das, D.; et al. Surface modification minimizes the toxicity of silver nanoparticles: An in vitro and in vivo study. J. Biol. Inorg. Chem. 2017, 22, 893–918.
- Choudhary, A.; Singh, S.; Ravichandiran, V. Toxicity, preparation methods and applications of silver nanoparticles: An update. Toxicol. Mech. Methods 2022, 32, 650–661.
- Li, J.; Chang, X.; Shang, M.; Niu, S.; Zhang, W.; Li, Y.; Sun, Z.; Wu, T.; Kong, L.; Zhang, T.; et al. The crosstalk between DRP1-dependent mitochondrial fission and oxidative stress triggers hepatocyte apoptosis induced by silver nanoparticles. Nanoscale 2021, 13, 12356–12369.
- Zhang, X.F.; Liu, Z.G.; Shen, W.; Gurunathan, S. Silver nanoparticles: Synthesis, characterization, properties, applications, and therapeutic approaches. Int. J. Mol. Sci. 2016, 17, 1534.
- Rohde, M.M.; Snyder, C.M.; Sloop, J.; Solst, S.R.; Donati, G.L.; Spitz, D.R.; Furdui, C.M.; Singh, R. The mechanism of cell death induced by silver nanoparticles is distinct from silver cations. Part. Fibre Toxicol. 2021, 18, 37.
- Wang, H.; Yan, A.; Liu, Z.; Yang, X.; Xu, Z.; Wang, Y.; Wang, R.; Koohi-Moghadam, M.; Hu, L.; Xia, W.; et al. Deciphering molecular mechanism of silver by integrated omic approaches enables enhancing its antimicrobial efficacy in E. coli. PLoS Biol. 2019, 17, e3000292.
- Elnaggar, M.G.; Jiang, K.; Eldesouky, H.E.; Pei, Y.; Park, J.; Yuk, S.A.; Meng, F.; Dieterly, A.M.; Mohammad, H.T.; Hegazy, Y.A.; et al. Antibacterial nanotruffles for treatment of intracellular bacterial infection. Biomaterials 2020, 262, 120344.
- Saris, N.E.; Skul’skiĭ, I.A.; Savina, M.V.; Glasunov, V.V. Mechanism of mitochondrial transport of thallous ions. J. Bioenerg. Biomembr. 1981, 13, 51–59.
- Skulskii, I.A.; Savina, M.V.; Glasunov, V.V.; Saris, N.E. Electrophoretic transport of Tl+ in mitochondria. J. Membr. Biol. 1978, 44, 187–194.
- Korotkov, S.M. Effects of Tl+ on ion permeability, membrane potential and respiration of isolated rat liver mitochondria. J. Bioenerg. Biomembr. 2009, 41, 277–287.
- Bragadin, M.; Toninello, A.; Bindoli, A.; Rigobello, M.P.; Canton, M. Thallium induces apoptosis in Jurkat cells. Ann. N. Y. Acad. Sci. 2003, 1010, 283–291.
- Korotkov, S.M.; Emel’yanova, L.V.; Yagodina, O.V. Inorganic phosphate stimulates the toxic effects of Tl+ in rat liver mitochondria. J. Biochem. Mol. Toxicol. 2008, 22, 148–157.
- Flesch, P.; Goldstone, S.B. Effect of thallium on sulphydryl compounds in vitro. J. Investig. Dermatol. 1950, 15, 345–347.
- Wojtovich, A.P.; Williams, D.M.; Karcz, M.K.; Lopes, C.M.; Gray, D.A.; Nehrke, K.W.; Brookes, P.S. A novel mitochondrial KATP channel assay. Circ. Res. 2010, 106, 1190–1196.
- Testai, L.; Martelli, A.; Marino, A.; D’antongiovanni, V.; Ciregia, F.; Giusti, L.; Lucacchini, A.; Chericoni, S.; Breschi, M.C.; Calderone, V. The activation of mitochondrial BK potassium channels contributes to the protective effects of naringenin against myocardial ischemia/reperfusion injury. Biochem. Pharmacol. 2013, 85, 1634–1643.
- Spencer, P.S.; Peterson, E.R.; Madrid, R.; Raine, C.S. Effects of thallium salts on neuronal mitochondria in organotypic cord-ganglia-muscle combination cultures. J. Cell Biol. 1973, 58, 79–95.
- Korotkov, S.M.; Saris, N.E. Influence of Tl+ on mitochondrial permeability transition pore in Ca2+-loaded rat liver mitochondria. J. Bioenerg. Biomembr. 2011, 43, 149–162.
- Korotkov, S.M.; Nesterov, V.P.; Brailovskaya, I.V.; Furaev, V.V.; Novozhilov, A.V. Tl+ induces both cationic and transition pore permeability in the inner membrane of rat heart mitochondria. J. Bioenerg. Biomembr. 2013, 45, 531–539.
- Korotkov, S.M.; Lapin, A.V. Thallium induces opening of the mitochondrial permeability transition pore in the inner membrane of rat liver mitochondria. Dokl. Biochem. Biophys. 2003, 392, 247–252.
- Korotkov, S.M.; Konovalova, S.A.; Brailovskaya, I.V.; Saris, N.E. To involvement the conformation of the adenine nucleotide translocase in opening the Tl+-induced permeability transition pore in Ca2+-loaded rat liver mitochondria. Toxicol. In Vitro 2016, 32, 320–332.
- Korotkov, S.; Konovalova, S.; Emelyanova, L.; Brailovskaya, I. Y3+, La3+, and some bivalent metals inhibited the opening of the Tl+-induced permeability transition pore in Ca2+-loaded rat liver mitochondria. J. Inorg. Biochem. 2014, 141, 1–9.
- Korotkov, S.M.; Brailovskaya, I.V.; Shumakov, A.R.; Emelyanova, L.V. Closure of mitochondrial potassium channels favors opening of the Tl+-induced permeability transition pore in Ca2+-loaded rat liver mitochondria. J. Bioenerg. Biomembr. 2015, 47, 243–254.
- Fontaine, E.; Eriksson, O.; Ichas, F.; Bernardi, P. Regulation of the permeability transition pore in skeletal muscle mitochondria. Modulation By electron flow through the respiratory chain complex I. J. Biol. Chem. 1998, 273, 12662–12668.
- Kowaltowski, A.J.; Vercesi, A.E.; Castilho, R.F. Mitochondrial membrane protein thiol reactivity with N-ethylmaleimide or mersalyl is modified by Ca2+: Correlation with mitochondrial permeability transition. Biochim. Biophys. Acta 1997, 1318, 395–402.
- McStay, G.P.; Clarke, S.J.; Halestrap, A.P. Role of critical thiol groups on the matrix surface of the adenine nucleotide translocase in the mechanism of the mitochondrial permeability transition pore. Biochem. J. 2002, 367, 541–548.
- Korotkov, S.M.; Konovalova, S.A.; Brailovskaya, I.V. Diamide accelerates opening of the Tl+-induced permeability transition pore in Ca2+-loaded rat liver mitochondria. Biochem. Biophys. Res. Commun. 2015, 468, 360–364.
- Korotkov, S.M.; Novozhilov, A.V. The joint influence of Tl+ and thiol-modifying agents on rat liver mitochondrial parameters in vitro. Int. J. Mol. Sci. 2022, 23, 8964.
- Korotkov, S.M.; Novozhilov, A.V. comparative study on the effects of the lysine reagent pyridoxal 5-phosphate and some thiol reagents in opening the Tl+-induced mitochondrial permeability transition pore. Int. J. Mol. Sci. 2023, 24, 2460.
- Hanzel, C.E.; Verstraeten, S.V. Thallium induces hydrogen peroxide generation by impairing mitochondrial function. Toxicol. Appl. Pharmacol. 2006, 216, 485–492.
- Hanzel, C.E.; Verstraeten, S.V. Tl(I) and Tl(III) activate both mitochondrial and extrinsic pathways of apoptosis in rat pheochromocytoma (PC12) cells. Toxicol. Appl. Pharmacol. 2009, 236, 59–70.
- Verstraeten, S.V. Relationship between thallium(I)-mediated plasma membrane fluidification and cell oxidants production in Jurkat T cells. Toxicology 2006, 222, 95–102.
- Pourahmad, J.; Eskandari, M.R.; Daraei, B. A comparison of hepatocyte cytotoxic mechanisms for thallium (I) and thallium (III). Environ. Toxicol. 2010, 25, 456–467.
- Eskandari, M.R.; Pourahmad, J.; Daraei, B. Thallium(I) and thallium(III) induce apoptosis in isolated rat hepatocytes by alterations in mitochondrial function and generation of ROS. Toxicol. Environ. Chem. 2011, 93, 145–156.
- Pino, M.T.; Marotte, C.; Verstraeten, S.V. Epidermal growth factor prevents thallium(I)- and thallium(III)-mediated rat pheochromocytoma (PC12) cell apoptosis. Arch. Toxicol. 2017, 91, 1157–1174.
- Maya-López, M.; Mireles-García, M.V.; Ramírez-Toledo, M.; Colín-González, A.L.; Galván-Arzate, S.; Túnez, I.; Santamaría, A. Thallium-induced toxicity in rat brain crude synaptosomal/mitochondrial fractions is sensitive to anti-excitatory and antioxidants agents. Neurotox. Res. 2018, 33, 634–640.
- Bramanti, E.; Onor, M.; Colombaioni, L. Neurotoxicity Induced by Low Thallium Doses in Living Hippocampal Neurons: Evidence of Early Onset Mitochondrial Dysfunction and Correlation with Ethanol Production. ACS Chem. Neurosci. 2019, 10, 451–459.
- Puga Molina, L.C.; Salvatierra Fréchou, D.M.; Verstraeten, S.V. Early response of glutathione- and thioredoxin-dependent antioxidants defense systems to Tl(I)- and Tl(III)-mediated oxidative stress in adherent pheochromocytoma (PC12adh) cells. Arch. Toxicol. 2017, 92, 195–211.
- Osorio-Rico, L.; Villeda-Hernández, J.; Santamaría, A.; Königsberg, M.; Galván-Arzate, S. The N-Methyl-d-aspartate receptor antagonist MK-801 prevents thallium-induced behavioral and biochemical alterations in the rat brain. Int. J. Toxicol. 2015, 34, 505–513.
- Belyaeva, E.A.; Glazunov, V.V.; Korotkov, S.M. Cd2+-promoted mitochondrial permeability transition: A comparison with other heavy metals. Acta Biochim. Pol. 2004, 51, 545–551.
- Ma, L.; Bi, K.D.; Fan, Y.M.; Jiang, Z.Y.; Zhang, X.Y.; Zhang, J.W.; Zhao, J.; Jiang, F.L.; Dong, J.X. In vitro modulation of mercury-induced rat liver mitochondria dysfunction. Toxicol. Res. 2018, 7, 1135–1143.
- Belyaeva, E.A. Modulators of mitochondrial ATP-sensitive potassium channel affect cytotoxicity of heavy metals: Action on isolated rat liver mitochondria and AS-30D ascites hepatoma cells. Ecotoxicol. Environ. Saf. 2023, 256, 114829.
- Ajsuvakova, O.P.; Tinkov, A.A.; Aschner, M.; Rocha, J.B.T.; Michalke, B.; Skalnaya, M.G.; Skalny, A.V.; Butnariu, M.; Dadar, M.; Sarac, I.; et al. Sulfhydryl groups as targets of mercury toxicity. Coord. Chem. Rev. 2020, 417, 213343.
- Nesci, S.; Trombetti, F.; Pirini, M.; Ventrella, V.; Pagliarani, A. Mercury and protein thiols: Stimulation of mitochondrial F1FO-ATPase and inhibition of respiration. Chem. Biol. Interact. 2016, 260, 42–49.
- Franco, J.L.; Braga, H.C.; Stringari, J.; Missau, F.C.; Posser, T.; Mendes, B.G.; Leal, R.B.; Santos, A.R.; Dafre, A.L.; Pizzolatti, M.G.; et al. Mercurial-induced hydrogen peroxide generation in mouse brain mitochondria: Protective effects of quercetin. Chem. Res. Toxicol. 2007, 20, 1919–1926.
- Hernández-Esquivel, L.; Zazueta, C.; Buelna-Chontal, M.; Hernández-Reséndiz, S.; Pavón, N.; Chávez, E. Protective behavior of tamoxifen against Hg2+-induced toxicity on kidney mitochondria: In vitro and in vivo experiments. J. Steroid Biochem. Mol. Biol. 2011, 127, 345–350.
- Novo, J.P.; Martins, B.; Raposo, R.S.; Pereira, F.C.; Oriá, R.B.; Malva, J.O.; Fontes-Ribeiro, C. Cellular and molecular mechanisms mediating methylmercury neurotoxicity and neuroinflammation. Int. J. Mol. Sci. 2021, 22, 3101.
- Palanirajan, S.K.; Gummadi, S.N. Heavy-Metals-Mediated Phospholipids Scrambling by Human Phospholipid Scramblase 3: A Probable Role in Mitochondrial Apoptosis. Chem. Res. Toxicol. 2020, 33, 553–564.
- Branco, V.; Godinho-Santos, A.; Gonçalves, J.; Lu, J.; Holmgren, A.; Carvalho, C. Mitochondrial thioredoxin reductase inhibition, selenium status, and Nrf-2 activation are determinant factors modulating the toxicity of mercury compounds. Free Radic. Biol. Med. 2014, 73, 95–105.
- Issa, Y.; Brunton, P.; Waters, C.M.; Watts, D.C. Cytotoxicity of metal ions to human oligodendroglial cells and human gingival fibroblasts assessed by mitochondrial dehydrogenase activity. Dent. Mater. 2008, 24, 281–287.
- Branco, V.; Coppo, L.; Solá, S.; Lu, J.; Rodrigues, C.M.P.; Holmgren, A.; Carvalho, C. Impaired cross-talk between the thioredoxin and glutathione systems is related to ASK-1 mediated apoptosis in neuronal cells exposed to mercury. Redox Biol. 2017, 13, 278–287.
- Chen, Y.W.; Huang, C.F.; Yang, C.Y.; Yen, C.C.; Tsai, K.S.; Liu, S.H. Inorganic mercury causes pancreatic beta-cell death via the oxidative stress-induced apoptotic and necrotic pathways. Toxicol. Appl. Pharmacol. 2010, 243, 323–331.
- Orr, S.E.; Barnes, M.C.; Joshee, L.; Uchakian, O.; McKallip, R.J.; Bridges, C.C. Potential mechanisms of cellular injury following exposure to a physiologically relevant species of inorganic mercury. Toxicol. Lett. 2019, 304, 13–20.
- Belyaeva, E.A.; Dymkowska, D.; Wieckowski, M.R.; Wojtczak, L. Mitochondria as an important target in heavy metal toxicity in rat hepatoma AS-30D cells. Toxicol. Appl. Pharmacol. 2008, 231, 34–42.
- Belyaeva, E.A.; Dymkowska, D.; Wieckowski, M.R.; Wojtczak, L. Reactive oxygen species produced by the mitochondrial respiratory chain are involved in Cd2+-induced injury of rat ascites hepatoma AS-30D cells. Biochim. Biophys. Acta 2006, 1757, 1568–1574.
- Jorissen, A.; Plum, L.M.; Rink, L.; Haase, H. Impact of lead and mercuric ions on the interleukin-2-dependent proliferation and survival of T cells. Arch. Toxicol. 2013, 87, 249–258.
- Cordier, W.; Yousaf, M.; Nell, M.J.; Steenkamp, V. Underlying mechanisms of cytotoxicity in HepG2 hepatocarcinoma cells exposed to arsenic, cadmium and mercury individually and in combination. Toxicol. In Vitro 2021, 72, 105101.
- Paesano, L.; Marmiroli, M.; Bianchi, M.G.; White, J.C.; Bussolati, O.; Zappettini, A.; Villani, M.; Marmiroli, N. Differences in toxicity, mitochondrial function and miRNome in human cells exposed in vitro to Cd as CdS quantum dots or ionic Cd. J. Hazard. Mater. 2020, 393, 122430.
- Yuan, L.; Shi, X.; Tang, B.Z.; Wang, W.X. Real-time in vitro monitoring of the subcellular toxicity of inorganic Hg and methylmercury in zebrafish cells. Aquat. Toxicol. 2021, 236, 105859.
- Liu, C.B.; Qu, G.B.; Cao, M.X.; Liang, Y.; Hu, L.G.; Shi, J.B.; Cai, Y.; Jiang, G.B. Distinct toxicological characteristics and mechanisms of Hg2+ and MeHg in Tetrahymena under low concentration exposure. Aquat. Toxicol. 2017, 193, 152–159.
- Agarwala, S.; Rao, N.B.; Mudholkar, K.; Bhuwania, R.; Satish Rao, B.S. Mangiferin, a dietary xanthone protects against mercury-induced toxicity in HepG2 cells. Environ. Toxicol. 2012, 27, 117–127.
- Stacchiotti, A.; Morandini, F.; Bettoni, F.; Schena, I.; Lavazza, A.; Grigolato, P.G.; Apostoli, P.; Rezzani, R.; Aleo, M.F. Stress proteins and oxidative damage in a renal derived cell line exposed to inorganic mercury and lead. Toxicology 2009, 264, 215–224.
- Yeh, J.H.; Chung, H.M.; Ho, C.M.; Jan, C.R. Mercury-induced Ca2+ increase and cytotoxicity in renal tubular cells. Life Sci. 2004, 74, 2075–2083.
- Pan, J.; Wei, Y.; Ni, L.; Li, X.; Deng, Y.; Xu, B.; Yang, T.; Sun, J.; Liu, W. Unbalanced ER-mitochondrial calcium homeostasis promotes mitochondrial dysfunction and associated apoptotic pathways activation in methylmercury exposed rat cortical neurons. J. Biochem. Mol. Toxicol. 2022, 36, e23136.
- Belyaeva, E.A.; Korotkov, S.M. Mechanism of primary Cd2+-induced rat liver mitochondria dysfunction: Discrete modes of Cd2+ action on calcium and thiol-dependent domains. Toxicol. Appl. Pharmacol. 2003, 192, 56–68.
- Ciapaite, J.; Nauciene, Z.; Baniene, R.; Wagner, M.J.; Krab, K.; Mildaziene, V. Modular kinetic analysis reveals differences in Cd2+ and Cu2+ ion-induced impairment of oxidative phosphorylation in liver. FEBS J. 2009, 276, 3656–3668.
- Chávez, E.; Briones, R.; Michel, B.; Bravo, C.; Jay, D. Evidence for the involvement of dithiol groups in mitochondrial calcium transport: Studies with cadmium. Arch. Biochem. Biophys. 1985, 242, 493–497.
- Pavón, N.; Buelna-Chontal, M.; Macías-López, A.; Correa, F.; Uribe-Álvarez, C.; Hernández-Esquivel, L.; Chávez, E. On the oxidative damage by cadmium to kidney mitochondrial functions. Biochem. Cell Biol. 2019, 97, 187–192.
- Zazueta, C.; Sánchez, C.; García, N.; Correa, F. Possible involvement of the adenine nucleotide translocase in the activation of the permeability transition pore induced by cadmium. Int. J. Biochem. Cell Biol. 2000, 32, 1093–1101.
- Lee, W.K.; Bork, U.; Gholamrezaei, F.; Thévenod, F. Cd2+-induced cytochrome c release in apoptotic proximal tubule cells: Role of mitochondrial permeability transition pore and Ca2+ uniporter. Am. J. Physiol. Ren. Physiol. 2005, 288, F27–F39.
- Zhang, Y.; Li, J.H.; Liu, X.R.; Jiang, F.L.; Tian, F.F.; Liu, Y. Spectroscopic and microscopic studies on the mechanisms of mitochondrial toxicity induced by different concentrations of cadmium. J. Membr. Biol. 2011, 241, 39–49.
- Li, M.; Xia, T.; Jiang, C.S.; Li, L.J.; Fu, J.L.; Zhou, Z.C. Cadmium directly induced the opening of membrane permeability pore of mitochondria which possibly involved in cadmium-triggered apoptosis. Toxicology 2003, 194, 19–33.
- Ardehali, H.; O’Rourke, B. Mitochondrial KATP channels in cell survival and death. J. Mol. Cell. Cardiol. 2005, 39, 7–16.
- Costa, A.D.; Jakob, R.; Costa, C.L.; Andrukhiv, K.; West, I.C.; Garlid, K.D. The mechanism by which the mitochondrial ATP-sensitive K+ channel opening and H2O2 inhibit the mitochondrial permeability transition. J. Biol. Chem. 2006, 281, 20801–20808.
- Kupsch, K.; Parvez, S.; Siemen, D.; Wolf, G. Modulation of the permeability transition pore by inhibition of the mitochondrial KATP channel in liver vs brain mitochondria. J. Membr. Biol. 2007, 215, 69–74.
- Belyaeva, E.A.; Glazunov, V.V.; Korotkov, S.M. Cd2+ versus Ca2+-produced mitochondrial membrane permeabilization: A proposed direct participation of respiratory complexes I and III. Chem. Biol. Interact. 2004, 150, 253–370.
- Thévenod, F. Catch me if you can! Novel aspects of cadmium transport in mammalian cells. Biometals 2010, 23, 857–875.
- Thévenod, F.; Fels, J.; Lee, W.K.; Zarbock, R. Channels.; transporters and receptors for cadmium and cadmium complexes in eukaryotic cells: Myths and facts. Biometals 2019, 32, 469–489.
- Choong, G.; Liu, Y.; Templeton, D.M. Interplay of calcium and cadmium in mediating cadmium toxicity. Chem. Biol. Interact. 2014, 211, 54–65.
- Thévenod, F.; Lee, W.K. Cadmium and cellular signaling cascades: Interactions between cell death and survival pathways. Arch. Toxicol. 2013, 87, 1743–1786.
- Yeh, J.H.; Huang, C.C.; Yeh, M.Y.; Wang, J.S.; Lee, J.K.; Jan, C.R. Cadmium-induced cytosolic Ca2+ elevation and subsequent apoptosis in renal tubular cells. Basic Clin. Pharmacol. Toxicol. 2009, 104, 345–351.
- Yang, C.S.; Tzou, B.C.; Liu, Y.P.; Tsai, M.J.; Shyue, S.K.; Tzeng, S.F. Inhibition of cadmium-induced oxidative injury in rat primary astrocytes by the addition of antioxidants and the reduction of intracellular calcium. J. Cell. Biochem. 2008, 103, 825–834.
- Wang, X.; Yi, M.; Liu, H.; Han, Y.; Yi, H. Reactive oxygen species and Ca2+ are involved in cadmium-induced cell killing in yeast cells. Can. J. Microbiol. 2017, 63, 153–159.
- Wang, H.; Yu, Y.; Li, J.; Wu, H.; Sun, J.; Zhang, Z.; Geng, L.; Yu, X.; Liu, Z. Cadmium stimulates mouse skin fibroblast apoptosis by affecting intracellular homeostasis. Drug Chem. Toxicol. 2017, 40, 74–84.
- Pourahmad, J.; O’Brien, P.J. Contrasting role of Na+ ions in modulating Cu2+ or Cd2+ induced hepatocyte toxicity. Chem. Biol. Interact. 2000, 126, 159–169.
- Yuan, Y.; Jiang, C.Y.; Xu, H.; Sun, Y.; Hu, F.F.; Bian, J.C.; Liu, X.Z.; Gu, J.H.; Liu, Z.P. Cadmium-induced apoptosis in primary rat cerebral cortical neurons culture is mediated by a calcium signaling pathway. PLoS ONE 2013, 8, e64330.
- Biagioli, M.; Pifferi, S.; Ragghianti, M.; Bucci, S.; Rizzuto, R.; Pinton, P. Endoplasmic reticulum stress and alteration in calcium homeostasis are involved in cadmium-induced apoptosis. Cell Calcium 2008, 43, 184–195.
- Lee, W.K.; Thévenod, F. Novel roles for ceramides, calpains and caspases in kidney proximal tubule cell apoptosis: Lessons from in vitro cadmium toxicity studies. Biochem. Pharmacol. 2008, 76, 1323–1332.
- Wätjen, W.; Cox, M.; Biagioli, M.; Beyersmann, D. Cadmium-induced apoptosis in C6 glioma cells: Mediation by caspase 9-activation. Biometals 2002, 15, 15–25.
- Wätjen, W.; Beyersmann, D. Cadmium-induced apoptosis in C6 glioma cells: Influence of oxidative stress. Biometals 2004, 17, 65–78.
- Zhang, C.; Yuan, X.; Mao, W.; Yue, L.; Kong, X.; Gao, Y.; Luo, L.; Yin, Z. Inhibition of cadmium-induced apoptosis by glutathione S-transferase P1 via mitogen-activated protein kinases, and mitochondrial pathways. Environ. Toxicol. Pharmacol. 2010, 30, 202–208.
- Murugavel, P.; Pari, L.; Sitasawad, S.L.; Kumar, S.; Kumar, S. Cadmium induced mitochondrial injury and apoptosis in vero cells: Protective effect of diallyl tetrasufide from garlic. Int. J. Biochem. Cell Biol. 2007, 39, 161–170.
- Kim, S.J.; Jeong, H.J.; Myung, N.Y.; Kim, M.C.; Lee, J.H.; So, H.S.; Park, R.K.; Kim, H.M.; Um, J.Y.; Hong, S.H. The protective mechanism of antioxidants in cadmium-induced ototoxicity in vitro and in vivo. Environ. Health Perspect. 2008, 116, 854–862.
- Nair, A.R.; Lee, W.K.; Smeets, K.; Swennen, Q.; Sanchez, A.; Thévenod, F.; Cuypers, A. Glutathione and mitochondria determine acute defense responses and adaptive processes in cadmium-induced oxidative stress and toxicity of the kidney. Arch. Toxicol. 2015, 89, 2273–2289.
- Thévenod, F. Nephrotoxicity and the proximal tubule. Insights from cadmium. Nephron Physiol. 2003, 93, p87–p93.
- Almazan, G.; Liu, H.N.; Khorchid, A.; Sundararajan, S.; Martinez-Bermudez, A.K.; Chemtob, S. Exposure of developing oligodendrocytes to cadmium causes HSP72 induction, free radical generation, reduction in glutathione levels, and cell death. Free Radic. Biol. Med. 2000, 29, 858–869.
- Pourahmad, J.; O’Brien, P.J. A comparison of hepatocyte cytotoxic mechanisms for Cu2+ and Cd2+. Toxicology 2000, 143, 263–273.
- Son, Y.O.; Lee, J.C.; Hitron, J.A.; Pan, J.; Zhang, Z.; Shi, X. Cadmium induces intracellular Ca2+- and H2O2-dependent apoptosis through JNK- and p53-mediated pathways in skin epidermal cell line. Toxicol. Sci. 2010, 113, 127–137.
- Pourahmad, J.; Mihajlovic, A.; O’Brien, P.J. Hepatocyte lysis induced by environmental metal toxins may involve apoptotic death signals initiated by mitochondrial injury. Adv. Exp. Med. Biol. 2001, 500, 249–252.
- Belyaeva, E.A.; Sokolova, T.V.; Emelyanova, L.V.; Zakharova, I.O. Mitochondrial electron transport chain in heavy metal-induced neurotoxicity: Effects of cadmium, mercury, and copper. Sci. World J. 2012, 2012, 136063.
- Belyaeva, E.A. Role of mitochondrial respiratory chain in neurotoxic action of heavy metals: Comparison of Cd2+, Hg2+ and Cu2+. Cut. Edge Res. Biol. 2022, 2, 144–174.
- Zapór, L. Evaluation of the toxic potency of selected cadmium compounds on A549 and CHO-9 cells. Int. J. Occup. Saf. Ergon. 2014, 20, 573–581.
- Sancho, P.; Fernández, C.; Yuste, V.J.; Amrán, D.; Ramos, A.M.; de Blas, E.; Susin, S.A.; Aller, P. Regulation of apoptosis/necrosis execution in cadmium-treated human promonocytic cells under different forms of oxidative stress. Apoptosis 2006, 11, 673–686.
- Mahdavi, S.; Khodarahmi, P.; Roodbari, N.H. Effects of cadmium on Bcl-2/ Bax expression ratio in rat cortex brain and hippocampus. Hum. Exp. Toxicol. 2018, 37, 321–328.
- Rikans, L.E.; Yamano, T. Mechanisms of cadmium-mediated acute hepatotoxicity. J. Biochem. Mol. Toxicol. 2000, 14, 110–117.
- Liu, Y.; Templeton, D.M. Role of the cytoskeleton in Cd2+-induced death of mouse mesangial cells. Can. J. Physiol. Pharmacol. 2010, 88, 341–352.
- Xiao, W.; Liu, Y.; Templeton, D.M. Pleiotropic effects of cadmium in mesangial cells. Toxicol. Appl. Pharmacol. 2009, 238, 315–326.
- Pillai, A.; Gupta, S. Antioxidant enzyme activity and lipid peroxidation in liver of female rats co-exposed to lead and cadmium: Effects of vitamin E and Mn2+. Free Radic. Res. 2005, 39, 707–712.
- Shih, C.M.; Ko, W.C.; Wu, J.S.; Wei, Y.H.; Wang, L.F.; Chang, E.E.; Lo, T.Y.; Cheng, H.H.; Chen, C.T. Mediating of caspase-independent apoptosis by cadmium through the mitochondria-ROS pathway in MRC-5 fibroblasts. J. Cell. Biochem. 2004, 91, 384–397.
- Tandon, S.K.; Singh, S.; Prasad, S.; Mathur, N. Hepatic and renal metallothionein induction by. an oral equimolar dose of zinc, cadmium or mercury in mice. Food Chem. Toxicol. 2001, 39, 571–577.
- Binte Hossain, K.F.; Rahman, M.M.; Sikder, M.T.; Saito, T.; Hosokawa, T.; Kurasaki, M. Inhibitory effects of selenium on cadmium-induced cytotoxicity in PC12 cells via regulating oxidative stress and apoptosis. Food Chem. Toxicol. 2018, 114, 180–189.
- Isaev, N.K.; Avilkina, S.; Golyshev, S.A.; Genrikhs, E.E.; Alexandrova, O.P.; Kapkaeva, M.R.; Stelmashook, E.V. N-acetyl-l-cysteine and Mn2+ attenuate Cd2+-induced disturbance of the intracellular free calcium homeostasis in cultured cerebellar granule neurons. Toxicology 2018, 393, 1–8.
- Stelmashook, E.V.; Alexandrova, O.P.; Genrikhs, E.E.; Novikova, S.V.; Salmina, A.B.; Isaev, N.K. Effect of zinc and copper ions on cadmium-induced toxicity in rat cultured cortical neurons. J. Trace Elem. Med. Biol. 2022, 73, 127012.
- Zhang, D.; Zhang, T.; Liu, J.; Chen, J.; Li, Y.; Ning, G.; Huo, N.; Tian, W.; Ma, H. Zn Supplement-Antagonized Cadmium-Induced Cytotoxicity in Macrophages In Vitro: Involvement of Cadmium Bioaccumulation and Metallothioneins Regulation. J. Agric. Food Chem. 2019, 67, 4611–4622.
- Rahman, M.M.; Ukiana, J.; Uson-Lopez, R.; Sikder, M.T.; Saito, T.; Kurasaki, M. Cytotoxic effects of cadmium and zinc co-exposure in PC12 cells and the underlying mechanism. Chem. Biol. Interact. 2017, 269, 41–49.
- Zhang, D.; Liu, J.; Gao, J.; Shahzad, M.; Han, Z.; Wang, Z.; Li, J.; Sjölinder, H. Zinc supplementation protects against cadmium accumulation and cytotoxicity in Madin-Darby bovine kidney cells. PLoS ONE 2014, 9, e103427.
- Barbier, O.; Dauby, A.; Jacquillet, G.; Tauc, M.; Poujeol, P.; Cougnon, M. Zinc and cadmium interactions in a renal cell line derived from rabbit proximal tubule. Nephron Physiol. 2005, 99, p74–p84.
- Poliandri, A.H.; Velardez, M.O.; Cabilla, J.P.; Bodo, C.C.; Machiavelli, L.I.; Quinteros, A.F.; Duvilanski, B.H. Nitric oxide protects anterior pituitary cells from cadmium-induced apoptosis. Free Radic. Biol. Med. 2004, 37, 1463–1471.
- Che, L.; Yang, C.L.; Chen, Y.; Wu, Z.L.; Du, Z.B.; Wu, J.S.; Gan, C.L.; Yan, S.P.; Huang, J.; Guo, N.J.; et al. Mitochondrial redox-driven mitofusin 2 S-glutathionylation promotes neuronal necroptosis via disrupting ER-mitochondria crosstalk in cadmium-induced neurotoxicity. Chemosphere 2021, 262, 127878.
- Ma, L.; Liu, J.Y.; Dong, J.X.; Xiao, Q.; Zhao, J.; Jiang, F.L. Toxicity of Pb2+ on rat liver mitochondria induced by oxidative stress and mitochondrial permeability transition. Toxicol. Res. 2017, 6, 822–830.
- Han, Q.; Zhang, W.; Guo, J.; Zhu, Q.; Chen, H.; Xia, Y.; Zhu, G. Mitochondrion: A sensitive target for Pb exposure. J. Toxicol. Sci. 2021, 46, 345–358.
- Yang, X.; Wang, B.; Zeng, H.; Cai, C.; Hu, Q.; Cai, S.; Xu, L.; Meng, X.; Zou, F. Role of the mitochondrial Ca2+ uniporter in Pb2+-induced oxidative stress in human neuroblastoma cells. Brain Res. 2014, 1575, 12–21.
- Ye, F.; Li, X.; Li, F.; Li, J.; Chang, W.; Yuan, J.; Chen, J. Cyclosporin A protects against Lead neurotoxicity through inhibiting mitochondrial permeability transition pore opening in nerve cells. Neurotoxicology 2016, 57, 203–213.
- Liu, G.; Li, Z.; Wang, J.; Wang, H.; Wang, Z.; Wang, L. Puerarin protects against lead-induced cytotoxicity in cultured primary rat proximal tubular cells. Hum. Exp. Toxicol. 2014, 33, 1071–1080.
- Liu, G.; Wang, Z.K.; Wang, Z.Y.; Yang, D.B.; Liu, Z.P.; Wang, L. Mitochondrial permeability transition and its regulatory components are implicated in apoptosis of primary cultures of rat proximal tubular cells exposed to lead. Arch. Toxicol. 2016, 90, 1193–1209.
- Liu, Z.; Wang, H.; Zhang, W.; Yuan, Z.; Yuan, H.; Liu, X.; Zhang, M.; Guo, X.; Guan, W. Lead induces Siberian tiger fibroblast apoptosis by interfering with intracellular homeostasis. Drug Chem. Toxicol. 2018, 41, 188–198.
- Daniel, P.V.; Kamthan, M.; Thakur, S.; Mondal, P. Molecular pathways dysregulated by Pb2+ exposure prompts pancreatic beta-cell dysfunction. Toxicol. Res. 2022, 11, 206–214.
- Guo, S.; Zhou, J.; Chen, X.; Yu, Y.; Ren, M.; Hu, G.; Liu, Y.; Zou, F. Bystander effects of PC12 cells treated with Pb2+ depend on ROS-mitochondria-dependent apoptotic signaling via gap-junctional intercellular communication. Toxicol. Lett. 2014, 229, 150–157.
- Agarwal, S.; Roy, S.; Ray, A.; Mazumder, S.; Bhattacharya, S. Arsenic trioxide and lead acetate induce apoptosis in adult rat hepatic stem cells. Cell Biol. Toxicol. 2009, 25, 403–413.
- Virgolini, M.B.; Aschner, M. Molecular mechanisms of lead neurotoxicity. Adv. Neurotoxicol. 2021, 5, 159–213.
- Elmorsy, E.; Al-Ghafari, A.; Al Doghaither, H.; Ghulam, J. Effects of environmental metals on mitochondrial bioenergetics of the CD-1 mice pancreatic beta-cells. Toxicol. In Vitro 2021, 70, 105015.
- Cai, P.; Zhu, Q.; Cao, Q.; Bai, Y.; Zou, H.; Gu, J.; Yuan, Y.; Liu, X.; Liu, Z.; Bian, J. Quercetin and Allicin Can Alleviate the Hepatotoxicity of Lead (Pb) through the PI3K Signaling Pathway. J. Agric. Food Chem. 2021, 69, 9451–9460.
- Struzyńska, L.; Chalimoniuk, M.; Sulkowski, G. The role of astroglia in Pb-exposed adult rat brain with respect to glutamate toxicity. Toxicology 2005, 212, 185–194.
- Parida, L.; Patel, T.N. Systemic impact of heavy metals and their role in cancer development: A review. Environ. Monit. Assess. 2023, 195, 766.
- Bjorklund, G.; Stejskal, V.; Urbina, M.A.; Dadar, M.; Chirumbolo, S.; Mutter, J. Metals and Parkinson’s Disease: Mechanisms and Biochemical Processes. Curr. Med. Chem. 2018, 25, 2198–2214.
- Wu, X.; Cobbina, S.J.; Mao, G.; Xu, H.; Zhang, Z.; Yang, L. A review of toxicity and mechanisms of individual and mixtures of heavy metals in the environment. Environ. Sci. Pollut. Res. Int. 2016, 23, 8244–8259.
- Gandolfi, L.; Stella, M.P.; Zambenedetti, P.; Zatta, P. Aluminum alters intracellular calcium homeostasis in vitro. Biochim. Biophys. Acta 1998, 1406, 315–320.
- Arab-Nozari, M.; Zamani, E.; Latifi, A.; Shaki, F. Mitochondrial toxicity of aluminium nanoparticles in comparison to its ionic form on isolated rat brain mitochondria. Bratisl. Lek. Listy 2019, 120, 516–522.
- De Marchi, U.; Mancon, M.; Battaglia, V.; Ceccon, S.; Cardellini, P.; Toninello, A. Influence of reactive oxygen species production by monoamine oxidase activity on aluminum-induced mitochondrial permeability transition. Cell Mol. Life Sci. 2004, 61, 2664–2671.
- Yuan, L.; Zhang, J.; Liu, Y.; Zhao, J.; Jiang, F.; Liu, Y. Indium (III) induces isolated mitochondrial permeability transition by inhibiting proton influx and triggering oxidative stress. J. Inorg. Biochem. 2017, 177, 17–26.
- Willhite, C.C.; Karyakina, N.A.; Yokel, R.A.; Yenugadhati, N.; Wisniewski, T.M.; Arnold, I.M.; Momoli, F.; Krewski, D. Systematic review of potential health risks posed by pharmaceutical, occupational and consumer exposures to metallic and nanoscale aluminum, aluminum oxides, aluminum hydroxide and its soluble salts. Crit. Rev. Toxicol. 2014, 44, 1–80.
- Matyja, E. Aluminum enhances glutamate-mediated neurotoxicity in organotypic cultures of rat hippocampus. Folia Neuropathol. 2000, 38, 47–53.
- Yu, Q.; Zhu, K.; Ding, Y.; Han, R.; Cheng, D. Comparative study of aluminum (Al) speciation on apoptosis-promoting process in PC12 cells: Correlations between morphological characteristics and mitochondrial kinetic disorder. J. Inorg. Biochem. 2022, 232, 111835.
- Xu, F.; Ren, L.; Song, M.; Shao, B.; Han, Y.; Cao, Z.; Li, Y. Fas- and mitochondria-mediated signaling pathway involved in osteoblast apoptosis induced by AlCl3. Biol. Trace Elem. Res. 2018, 184, 173–185.
- Zhu, Y.; Xu, F.; Yan, X.; Miao, L.; Li, H.; Hu, C.; Wang, Z.; Lian, S.; Feng, Z.; Li, Y. The suppressive effects of aluminum chloride on the osteoblasts function. Environ. Toxicol. Pharmacol. 2016, 48, 125–129.
- Wang, X.; Fan, X.; Yuan, S.; Jiao, W.; Liu, B.; Cao, J.; Jiang, W. Chlorogenic acid protects against aluminium-induced cytotoxicity through chelation and antioxidant actions in primary hippocampal neuronal cells. Food Funct. 2017, 8, 2924–2934.
- Mailloux, R.; Lemire, J.; Appanna, V. Aluminum-induced mitochondrial dysfunction leads to lipid accumulation in human hepatocytes: A link to obesity. Cell. Physiol. Biochem. 2007, 20, 627–638.
- Zhang, Q.L.; Niu, P.Y.; Niu, Q.; Wang, L.P. Effect of aluminum on neuronal mitochondria of rats. Wei Sheng Yan Jiu. (J. Hyg. Res.) 2005, 34, 674–677. (In Chinese)
- Li, M.; Song, M.; Ren, L.M.; Xiu, C.Y.; Liu, J.Y.; Zhu, Y.Z.; Li, Y.F. AlCl3 induces lymphocyte apoptosis in rats through the mitochondria-caspase dependent pathway. Environ. Toxicol. 2016, 31, 385–394.
- Masson, J.D.; Badran, G.; Domdom, M.A.; Gherardi, R.K.; Mograbi, B.; Authier, F.J.; Crépeaux, G. Advances on the early cellular events occurring upon exposure of human macrophages to aluminum oxyhydroxide adjuvant. Sci. Rep. 2023, 13, 3198.
- Xu, F.; Liu, Y.; Zhao, H.; Yu, K.; Song, M.; Zhu, Y.; Li, Y. Aluminum chloride caused liver dysfunction and mitochondrial energy metabolism disorder in rat. J. Inorg. Biochem. 2017, 174, 55–62.
- Sieg, H.; Ellermann, A.L.; Maria Kunz, B.; Jalili, P.; Burel, A.; Hogeveen, K.; Böhmert, L.; Chevance, S.; Braeuning, A.; Gauffre, F.; et al. Aluminum in liver cells—The element species matters. Nanotoxicology 2019, 13, 909–922.
- Chitambar, C.R. Gallium Complexes as Anticancer Drugs. Met. Ions Life Sci. 2018, 18, 281–301.
- Ahamed, M.; Akhtar, M.J.; Khan, M.A.M.; Alhadlaq, H.A.; Aldalbahi, A. Nanocubes of indium oxide induce cytotoxicity and apoptosis through oxidative stress in human lung epithelial cells. Colloids Surf. B 2017, 156, 157–164.
- Alkahtane, A.A. Indium tin oxide nanoparticles-mediated DNA fragmentation and cell death by apoptosis in human lung epithelial cells. Toxicol. Environ. Chem. 2015, 97, 1086–1098.
- Badding, M.A.; Stefaniak, A.B.; Fix, N.R.; Cummings, K.J.; Leonard, S.S. Cytotoxicity and characterization of particles collected from an indium-tin oxide production facility. J. Toxicol. Environ. Health A 2014, 77, 1193–1209.
- Gwinn, W.M.; Qu, W.; Shines, C.J.; Bousquet, R.W.; Taylor, G.J.; Waalkes, M.P.; Morgan, D.L. Macrophage solubilization and cytotoxicity of indium-containing particles in vitro. Toxicol. Sci. 2013, 135, 414–424.
- Tabei, Y.; Sonoda, A.; Nakajima, Y.; Biju, V.; Makita, Y.; Yoshida, Y.; Horie, M. Intracellular accumulation of indium ions released from nanoparticles induces oxidative stress, proinflammatory response and DNA damage. J. Biochem. 2016, 159, 225–237.
- Eskandari, A.; Glerum, D.M.; Tsui, T.Y. Influence of Indium (III) Chloride on Human Dermal Fibroblast Cell Adhesion on Tantalum/Silicon Oxide Nano-Composites. Materials 2022, 15, 3577.
- Li, H.; Chen, Z.; Li, J.; Liu, R.; Zhao, F.; Liu, R. Indium oxide nanoparticles induce lung intercellular toxicity between bronchial epithelial cells and macrophages. J. Appl. Toxicol. 2020, 40, 1636–1646.
- Kalo, M.B.; Rezaei, M. In vitro toxic interaction of arsenic and hyperglycemia in mitochondria: An important implication of increased vulnerability in pre-diabetics. Environ. Sci. Pollut. Res. Int. 2022, 29, 28375–28385.
- Mozaffarian, F.; Dehghani, M.A.; Vanani, A.R.; Mahdavinia, M. Protective effects of alpha lipoic acid against arsenic induced oxidative stress in isolated rat liver mitochondria. Biol. Trace Elem. Res. 2022, 200, 1190–1200.
- Ahangarpour, A.; Zeidooni, L.; Rezaei, M.; Alboghobeish, S.; Samimi, A.; Oroojan, A.A. Protective effect of metformin on toxicity of butyric acid and arsenic in isolated liver mitochondria and langerhans islets in male mice: An in vitro study. Iran. J. Basic Med. Sci. 2017, 20, 1297–1305.
- Hosseini, M.J.; Shaki, F.; Ghazi-Khansari, M.; Pourahmad, J. Toxicity of Arsenic (III) on Isolated Liver Mitochondria: A New Mechanistic Approach. Iran. J. Pharm. Res. 2013, 12, 121–138.
- Keshtzar, E.; Khodayar, M.J.; Javadipour, M.; Ghaffari, M.A.; Bolduc, D.L.; Rezaei, M. Ellagic acid protects against arsenic toxicity in isolated rat mitochondria possibly through the maintaining of complex II. Hum. Exp. Toxicol. 2016, 35, 1060–1072.
- Rigobello, M.P.; Folda, A.; Scutari, G.; Bindoli, A. The modulation of thiol redox state affects the production and metabolism of hydrogen peroxide by heart mitochondria. Arch. Biochem. Biophys. 2005, 441, 112–122.
- Tian, X.; Ma, X.; Qiao, D.; Ma, A.; Yan, F.; Huang, X. mCICR is required for As2O3-induced permeability transition pore opening and cytochrome c release from mitochondria. Mol. Cell. Biochem. 2005, 277, 33–42.
- Bustamante, J.; Nutt, L.; Orrenius, S.; Gogvadze, V. Arsenic stimulates release of cytochrome c from isolated mitochondria via induction of mitochondrial permeability transition. Toxicol. Appl. Pharmacol. 2005, 207, S110–S116.
- Németi, B.; Gregus, Z. Mitochondria work as reactors in reducing arsenate to arsenite. Toxicol. Appl. Pharmacol. 2002, 182, 208–218.
- Wu, Q.; Lu, Y.F.; Shi, J.Z.; Liang, S.X.; Shi, J.S.; Liu, J. Chemical form of metals in traditional medicines underlines potential toxicity in cell cultures. J. Ethnopharmacol. 2011, 134, 839–843.
- Fiorani, M.; Guidarelli, A.; Capellacci, V.; Cerioni, L.; Crinelli, R.; Cantoni, O. The dual role of mitochondrial superoxide in arsenite toxicity: Signaling at the boundary between apoptotic commitment and cytoprotection. Toxicol. Appl. Pharmacol. 2018, 345, 26–35.
- Guidarelli, A.; Fiorani, M.; Cerioni, L.; Scotti, M.; Cantoni, O. Arsenite induces DNA damage via mitochondrial ROS and induction of mitochondrial permeability transition. Biofactors 2017, 43, 673–684.
- Guidarelli, A.; Fiorani, M.; Cantoni, O. Low concentrations of arsenite target the intraluminal inositol 1, 4, 5-trisphosphate receptor/ryanodine receptor crosstalk to significantly elevate intracellular Ca2+. J. Pharmacol. Exp. Ther. 2018, 367, 184–193.
- Chen, C.; Gu, S.; Jiang, X.; Zhang, Z. Arsenite-induced endoplasmic reticulum-dependent apoptosis through disturbance of calcium homeostasis in HBE cell line. Environ. Toxicol. 2017, 32, 197–216.
- King, Y.A.; Chiu, Y.J.; Chen, H.P.; Kuo, D.H.; Lu, C.C.; Yang, J.S. Endoplasmic reticulum stress contributes to arsenic trioxide-induced intrinsic apoptosis in human umbilical and bone marrow mesenchymal stem cells. Environ. Toxicol. 2016, 31, 314–328.
- Zhang, H.; He, Z.; Deng, P.; Lu, M.; Zhou, C.; Yang, L.; Yu, Z. PIN1-mediated ROS production is involved in antagonism of N-acetyl-L-cysteine against arsenic-induced hepatotoxicity. Toxicol. Res. 2022, 11, 628–643.
- Thompson, J.A.; Franklin, C.C. Enhanced glutathione biosynthetic capacity promotes resistance to As3+-induced apoptosis. Toxicol. Lett. 2010, 193, 33–40.
- Ran, S.; Gao, X.; Ma, M.; Zhang, J.; Li, S.; Zhang, M.; Li, S. NaAsO2 decreases GSH synthesis by inhibiting GCLC and induces apoptosis through Hela cell mitochondrial damage, mediating the activation of the NF-κB/miR-21 signaling pathway. Ecotoxicol. Environ. Saf. 2022, 234, 113380.
- Duan, T.; Hu, T.; Wu, C.; Yeh, Y.T.; Lu, J.; Zhang, Q.; Li, X.; Jian, W.; Luo, P. PINK1/Parkin-mediated mitophagy is involved in NaAsO2-induced apoptosis of human hepatic cells through activation of ERK signaling. Toxicol. In Vitro 2020, 66, 104857.
- Wei, S.; Qiu, T.; Yao, X.; Wang, N.; Jiang, L.; Jia, X.; Tao, Y.; Wang, Z.; Pei, P.; Zhang, J.; et al. Arsenic induces pancreatic dysfunction and ferroptosis via mitochondrial ROS-autophagy-lysosomal pathway. J. Hazard. Mater. 2020, 384, 121390.
- Cheng, B.; Yang, X.; An, L.; Gao, B.; Liu, X. Arsenic trioxide-induced apoptosis of Hep-2 cell line through modulating intracellular glutathione (GSH) level. Auris Nasus Larynx 2010, 37, 89–94.
- Wang, Y.; Xu, Y.; Wang, H.; Xue, P.; Li, X.; Li, B.; Zheng, Q.; Sun, G. Arsenic induces mitochondria-dependent apoptosis by reactive oxygen species generation rather than glutathione depletion in Chang human hepatocytes. Arch. Toxicol. 2009, 83, 899–908.
- Ray, A.; Roy, S.; Agarwal, S.; Bhattacharya, S. As2O3 toxicity in rat hepatocytes: Manifestation of caspase-mediated apoptosis. Toxicol. Ind. Health 2008, 24, 643–653.
- Tian, X.; Wang, M.; Ying, X.; Dong, N.; Li, M.; Feng, J.; Zhao, Y.; Zhao, Q.; Tian, F.; Li, B.; et al. Co-exposure to arsenic and fluoride to explore the interactive effect on oxidative stress and autophagy in myocardial tissue and cell. Ecotoxicol. Environ. Saf. 2023, 253, 114647.
- Stevens, J.J.; Graham, B.; Dugo, E.; Berhaneselassie-Sumner, B.; Ndebele, K.; Tchounwou, P.B. Arsenic trioxide induces apoptosis via specific signaling pathways in HT-29 colon cancer cells. J. Cancer Sci. Ther. 2017, 9, 298–306.
- Qi, Z.; Wang, Q.; Wang, H.; Tan, M. Metallothionein Attenuated Arsenic-Induced Cytotoxicity: The Underlying Mechanism Reflected by Metabolomics and Lipidomics. J. Agric. Food Chem. 2021, 69, 5372–5380.
- Pourahmad, J.; Rabiei, M.; Jokar, F.; O’brien, P.J. A comparison of hepatocyte cytotoxic mechanisms for chromate and arsenite. Toxicology 2005, 206, 449–460.
- Wang, L.; Liu, S.; Gao, C.; Chen, H.; Li, J.; Lu, J.; Yuan, Y.; Zheng, X.; He, H.; Zhang, X.; et al. Arsenic trioxide-induced cardiotoxicity triggers ferroptosis in cardiomyoblast cells. Hum. Exp. Toxicol. 2022, 41, 1–9.
- Wang, W.; Zhang, A. HIF-1α mediates arsenic-induced metabolic reprogramming in lung bronchial epithelial cells. Biol. Trace Elem. Res. 2023, 201, 2284–2293.
- Chavan, H.; Christudoss, P.; Mickey, K.; Tessman, R.; Ni, H.M.; Swerdlow, R.; Krishnamurthy, P. Arsenite effects on mitochondrial bioenergetics in human and mouse primary hepatocytes follow a nonlinear dose response. Oxid. Med. Cell. Longev. 2017, 2017, 9251303.
- Wu, P.J.; Hsin, I.L.; Hung, W.L.; Lee, M.S.; Wang, P.H.; Ko, J.L. Combination treatment with cyclosporin A and arsenic trioxide induce synergistic cell death via non-apoptotic pathway in uterine cervical cancer cells. Chem. Biol. Interact. 2022, 368, 110177.
- Santra, A.; Bishnu, D.; Santra, S.; Ghatak, S.; Mukherjee, P.S.; Dhali, G.K.; Chowdhury, A. Arsenic-induced injury of mouse hepatocytes through lysosome and mitochondria: An in vitro study. Int. J. Hepatol. 2022, 2022, 1546297.
- Larochette, N.; Decaudin, D.; Jacotot, E.; Brenner, C.; Marzo, I.; Susin, S.A.; Zamzami, N.; Xie, Z.; Reed, J.; Kroemer, G. Arsenite induces apoptosis via a direct effect on the mitochondrial permeability transition pore. Exp. Cell Res. 1999, 249, 413–421.
- Tang, Q.; Bai, L.; Zou, Z.; Meng, P.; Xia, Y.; Cheng, S.; Mu, S.; Zhou, J.; Wang, X.; Qin, X.; et al. Ferroptosis is newly characterized form of neuronal cell death in response to arsenite exposure. Neurotoxicology 2018, 67, 27–36.
- Costantini, P.; Belzacq, A.S.; Vieira, H.L.; Larochette, N.; de Pablo, M.A.; Zamzami, N.; Susin, S.A.; Brenner, C.; Kroemer, G. Oxidation of a critical thiol residue of the adenine nucleotide translocator enforces Bcl-2-independent permeability transition pore opening and apoptosis. Oncogene 2000, 19, 307–314.
- He, Z.; Zhang, Y.; Zhang, H.; Zhou, C.; Ma, Q.; Deng, P.; Lu, M.; Mou, Z.; Lin, M.; Yang, L.; et al. NAC antagonizes arsenic-induced neurotoxicity through TMEM179 by inhibiting oxidative stress in Oli-neu cells. Ecotoxicol. Environ. Saf. 2021, 223, 112554.
- Guidarelli, A.; Carloni, S.; Balduini, W.; Fiorani, M.; Cantoni, O. Mitochondrial ascorbic acid prevents mitochondrial O2.- formation, an event critical for U937 cell apoptosis induced by arsenite through both autophagic-dependent and independent mechanisms. Biofactors 2016, 42, 190–200.
- Wang, S.; Geng, Z.; Shi, N.; Li, X.; Wang, Z. Dose-dependent effects of selenite (Se4+) on arsenite (As3+)-induced apoptosis and differentiation in acute promyelocytic leukemia cells. Cell Death Dis. 2015, 6, e1596.
- Zhou, J.; Ci, X.; Ma, X.; Yu, Q.; Cui, Y.; Zhen, Y.; Li, S. Pterostilbene Activates the Nrf2-Dependent Antioxidant Response to Ameliorate Arsenic-Induced Intracellular Damage and Apoptosis in Human Keratinocytes. Front. Pharmacol. 2019, 10, 497.
- Duan, X.; Li, J.; Li, W.; Xing, X.; Zhang, Y.; Li, W.; Zhao, L.; Sun, G.; Gao, X.H.; Li, B. Antioxidant tert-butylhydroquinone ameliorates arsenic-induced intracellular damages and apoptosis through induction of Nrf2-dependent antioxidant responses as well as stabilization of anti-apoptotic factor Bcl-2 in human keratinocytes. Free Radic. Biol. Med. 2016, 94, 74–87.
- Yang, Y.P.; Liang, Z.Q.; Gao, B.; Jia, Y.L.; Qin, Z.H. Dynamic effects of autophagy on arsenic trioxide-induced death of human leukemia cell line HL60 cells. Acta Pharmacol. Sin. 2008, 29, 123–134.
- Zhang, H.; Jin, B.; Liu, L.; Li, H.; Zheng, X.; Li, M.; He, R.; Wang, K. Glutathione Might Attenuate Arsenic-Induced Liver Injury by Modulating the Foxa2-XIAP Axis to Reduce Oxidative Stress and Mitochondrial Apoptosis. Biol. Trace Elem. Res. 2023, 201, 5201–5212.
- Jahangirnejad, R.; Goudarzi, M.; Kalantari, H.; Najafzadeh, H.; Rezaei, M. Subcellular organelle toxicity caused by arsenic nanoparticles in isolated rat hepatocytes. Int. J. Occup. Environ. Med. 2020, 11, 41–52.
- Su, L.; Fang, W.; Zhao, X.; Zhu, L.; Gao, L.; Chen, G. Disruption of mitochondrial redox homeostasis as a mechanism of antimony-induced reactive oxygen species and cytotoxicity. Ecotoxicol. Environ. Saf. 2022, 237, 113519.
- Yamamoto, E.S.; de Jesus, J.A.; Bezerra-Souza, A.; Brito, J.R.; Lago, J.H.G.; Laurenti, M.D.; Passero, L.F.D. Tolnaftate inhibits ergosterol production and impacts cell viability of Leishmania sp. Bioorg. Chem. 2020, 102, 104056.
- Keogan, D.M.; Oliveira, S.S.C.; Sangenito, L.S.; Branquinha, M.H.; Jagoo, R.D.; Twamley, B.; Santos, A.L.S.; Griffith, D.M. Novel antimony(iii) hydroxamic acid complexes as potential anti-leishmanial agents. Dalton Trans. 2018, 47, 7245–7255.
- Andrade, J.M.; Gonçalves, L.O.; Liarte, D.B.; Lima, D.A.; Guimarães, F.G.; de Melo Resende, D.; Santi, A.M.M.; de Oliveira, L.M.; Velloso, J.P.L.; Delfino, R.G.; et al. Comparative transcriptomic analysis of antimony resistant and susceptible Leishmania infantum lines. Parasite Vectors 2020, 13, 600.
- Verdugo, M.; Ogra, Y.; Quiroz, W. Mechanisms underlying the toxic effects of antimony species in human embryonic kidney cells (HEK-293) and their comparison with arsenic species. J. Toxicol. Sci. 2016, 41, 783–792.
- Lösler, S.; Schlief, S.; Kneifel, C.; Thiel, E.; Schrezenmeier, H.; Rojewski, M.T. Antimony-trioxide- and arsenic-trioxide-induced apoptosis in myelogenic and lymphatic cell lines, recruitment of caspases, and loss of mitochondrial membrane potential are enhanced by modulators of the cellular glutathione redox system. Ann. Hematol. 2009, 88, 1047–1058.
- Lecureur, V.; Le Thiec, A.; Le Meur, A.; Amiot, L.; Drenou, B.; Bernard, M.; Lamy, T.; Fauchet, R.; Fardel, O. Potassium antimonyl tartrate induces caspase- and reactive oxygen species-dependent apoptosis in lymphoid tumoral cells. Br. J. Haematol. 2002, 119, 608–615.
- Lecureur, V.; Lagadic-Gossmann, D.; Fardel, O. Potassium antimonyl tartrate induces reactive oxygen species-related apoptosis in human myeloid leukemic HL60 cells. Int. J. Oncol. 2002, 20, 1071–1076.
- Xie, Y.; Zhong, C.; Zeng, M.; Guan, L.; Luo, L. Effect of hexavalent chromium on electron leakage of respiratory chain in mitochondria isolated from rat liver. Cell. Physiol. Biochem. 2013, 31, 473–485.
- Fernandes, M.A.; Santos, M.S.; Alpoim, M.C.; Madeira, V.M.; Vicente, J.A. Chromium(VI) interaction with plant and animal mitochondrial bioenergetics: A comparative study. J. Biochem. Mol. Toxicol. 2002, 16, 53–63.
- Rossi, S.C.; Gorman, N.; Wetterhahn, K.E. Mitochondrial reduction of the carcinogen chromate: Formation of chromium(V). Chem. Res. Toxicol. 1988, 1, 101–107.
- Xiao, F.; Feng, X.; Zeng, M.; Guan, L.; Hu, Q.; Zhong, C. Hexavalent chromium induces energy metabolism disturbance and p53-dependent cell cycle arrest via reactive oxygen species in L-02 hepatocytes. Mol. Cell. Biochem. 2012, 371, 65–76.
- Zhang, Y.; Yang, G.; Huang, S.; Yang, X.; Yuan, F.; Song, Y.; Liu, S.; Yu, X. Regulation of Cr(VI)-Induced Premature Senescence in L02 Hepatocytes by ROS-Ca2+-NF-κB Signaling. Oxid. Med. Cell. Longev. 2022, 2022, 7295224.
- Liang, N.; Li, S.; Liang, Y.; Ma, Y.; Tang, S.; Ye, S.; Xiao, F. Clusterin inhibits Cr(VI)-induced apoptosis via enhancing mitochondrial biogenesis through AKT-associated STAT3 activation in L02 hepatocytes. Ecotoxicol. Environ. Saf. 2021, 221, 112447.
- Zhang, X.; Wang, Y.; Chen, M.; Zeng, M. Hexavalent chromium-induced apoptosis in Hep3B cells is accompanied by calcium overload, mitochondrial damage, and AIF translocation. Ecotoxicol. Environ. Saf. 2021, 208, 111391.
- Zhang, Y.; Bian, H.; Ma, Y.; Xiao, Y.; Xiao, F. Cr(VI)-induced overactive mitophagy contributes to mitochondrial loss and cytotoxicity in L02 hepatocytes. Biochem. J. 2020, 477, 2607–2619.
- Zhang, Y.; Ma, Y.; Liang, N.; Liang, Y.; Lu, C.; Xiao, F. Blockage of ROS-ERK-DLP1 signaling and mitochondrial fission alleviates Cr(VI)-induced mitochondrial dysfunction in L02 hepatocytes. Ecotoxicol. Environ. Saf. 2019, 186, 109749.
- Liang, Q.; Zhang, Y.; Huang, M.; Xiao, Y.; Xiao, F. Role of mitochondrial damage in Cr(VI)-induced endoplasmic reticulum stress in L-02 hepatocytes. Mol. Med. Rep. 2019, 19, 1256–1265.
- Xiao, F.; Li, Y.; Luo, L.; Xie, Y.; Zeng, M.; Wang, A.; Chen, H.; Zhong, C. Role of mitochondrial electron transport chain dysfunction in Cr(VI)-induced cytotoxicity in L-02 hepatocytes. Cell. Physiol. Biochem. 2014, 33, 1013–1025.
- Xiao, F.; Li, Y.; Dai, L.; Deng, Y.; Zou, Y.; Li, P.; Yang, Y.; Zhong, C. Hexavalent chromium targets mitochondrial respiratory chain complex I to induce reactive oxygen species-dependent caspase-3 activation in L-02 hepatocytes. Int. J. Mol. Med. 2012, 30, 629–635.
- Xiao, J.W.; Zhong, C.G.; Li, B. Study of L-02 hepatocyte apoptosis induced by hexavalent chromium associated with mitochondria function damage. Wei Sheng Yan Jiu (J. Hyg. Res.) 2006, 35, 416–418. (In Chinese)
- Lv, Y.; Li, T.; Yang, M.; Su, L.; Zhu, Z.; Zhao, S.; Zeng, W.; Zheng, Y. Melatonin attenuates chromium(VI)-induced spermatogonial stem cell/progenitor mitophagy by restoration of METTL3-mediated RNA N6-methyladenosine modification. Front. Cell Dev. Biol. 2021, 9, 684398.
- Dlamini, M.B.; Gao, Z.; Hasenbilige; Jiang, L.; Geng, C.; Li, Q.; Shi, X.; Liu, Y.; Cao, J. The crosstalk between mitochondrial dysfunction and endoplasmic reticulum stress promoted ATF4-mediated mitophagy induced by hexavalent chromium. Environ. Toxicol. 2021, 36, 1162–1172.
- Tang, S.; Ye, S.; Ma, Y.; Liang, Y.; Liang, N.; Xiao, F. Clusterin alleviates Cr(VI)-induced mitochondrial apoptosis in L02 hepatocytes via inhibition of Ca2+-ROS-Drp1-mitochondrial fission axis. Ecotoxicol. Environ. Saf. 2020, 205, 111326.
- Fu, S.C.; Liu, J.M.; Lee, K.I.; Tang, F.C.; Fang, K.M.; Yang, C.Y.; Su, C.C.; Chen, H.H.; Hsu, R.J.; Chen, Y.W. Cr(VI) induces ROS-mediated mitochondrial-dependent apoptosis in neuronal cells via the activation of Akt/ERK/AMPK signaling pathway. Toxicol. In Vitro 2020, 65, 104795.
- Clementino, M.; Kim, D.; Zhang, Z. Constitutive activation of NAD-dependent sirtuin 3 plays an important role in tumorigenesis of chromium(VI)-transformed cells. Toxicol. Sci. 2019, 169, 224–234.
- Ge, H.; Li, Z.; Jiang, L.; Li, Q.; Geng, C.; Yao, X.; Shi, X.; Liu, Y.; Cao, J. Cr (VI) induces crosstalk between apoptosis and autophagy through endoplasmic reticulum stress in A549 cells. Chem. Biol. Interact. 2019, 298, 35–42.
- Dashti, A.; Soodi, M.; Amani, N. Cr (VI) induced oxidative stress and toxicity in cultured cerebellar granule neurons at different stages of development and protective effect of Rosmarinic acid. Environ. Toxicol. 2016, 31, 269–277.
- Zeng, M.; Xiao, F.; Zhong, X.; Jin, F.; Guan, L.; Wang, A.; Liu, X.; Zhong, C. Reactive oxygen species play a central role in hexavalent chromium-induced apoptosis in Hep3B cells without the functional roles of p53 and caspase-3. Cell. Physiol. Biochem. 2013, 32, 279–290.
- Banu, S.K.; Stanley, J.A.; Lee, J.; Stephen, S.D.; Arosh, J.A.; Hoyer, P.B.; Burghardt, R.C. Hexavalent chromium-induced apoptosis of granulosa cells involves selective sub-cellular translocation of Bcl-2 members, ERK1/2 and p53. Toxicol. Appl. Pharmacol. 2011, 251, 253–266.
- Son, Y.O.; Hitron, J.A.; Wang, X.; Chang, Q.; Pan, J.; Zhang, Z.; Liu, J.; Wang, S.; Lee, J.C.; Shi, X. Cr(VI) induces mitochondrial-mediated and caspase-dependent apoptosis through reactive oxygen species-mediated p53 activation in JB6 Cl41 cells. Toxicol. Appl. Pharmacol. 2010, 245, 226–235.
- Russo, P.; Catassi, A.; Cesario, A.; Imperatori, A.; Rotolo, N.; Fini, M.; Granone, P.; Dominioni, L. Molecular mechanisms of hexavalent chromium-induced apoptosis in human bronchoalveolar cells. Am. J. Respir. Cell Mol. Biol. 2005, 33, 589–600.
- Hayashi, Y.; Kondo, T.; Zhao, Q.L.; Ogawa, R.; Cui, Z.G.; Feril, L.B., Jr.; Teranishi, H.; Kasuya, M. Signal transduction of p53-independent apoptotic pathway induced by hexavalent chromium in U937 cells. Toxicol. Appl. Pharmacol. 2004, 197, 96–106.
- Rudolf, E.; Cervinka, M.; Cerman, J. Zinc has ambiguous effects on chromium (VI)-induced oxidative stress and apoptosis. J. Trace Elem. Med. Biol. 2005, 18, 251–260.
- Azad, N.; Iyer, A.K.; Wang, L.; Lu, Y.; Medan, D.; Castranova, V.; Rojanasakul, Y. Nitric oxide-mediated bcl-2 stabilization potentiates malignant transformation of human lung epithelial cells. Am. J. Respir. Cell Mol. Biol. 2010, 42, 578–585.
- Belosludtsev, K.N.; Garmash, S.A.; Belosludtseva, N.V.; Belova, S.P.; Berezhnov, A.V.; Gudkov, S.V. Study of the mechanisms of cytotoxic effect of uranyl nitrate. Biophysics 2012, 57, 607–612.
- Shaki, F.; Hosseini, M.J.; Ghazi-Khansari, M.; Pourahmad, J. Toxicity of depleted uranium on isolated rat kidney mitochondria. Biochim. Biophys. Acta 2012, 1820, 1940–1950.
- Pourahmad, J.; Ghashang, M.; Ettehadi, H.A.; Ghalandari, R. A search for cellular and molecular mechanisms involved indepleted uranium (DU) toxicity. Environ. Toxicol. 2006, 21, 349–354.
- Guéguen, Y.; Frerejacques, M. Review of knowledge of uranium-induced kidney toxicity for the development of an adverse outcome pathway to renal impairment. Int. J. Mol. Sci. 2022, 23, 4397.
- Daraie, B.; Pourahmad, J.; Hamidi-Pour, N.; Hosseini, M.J.; Shaki, F.; Soleimani, M. Uranyl acetate induces oxidative stress and mitochondrial membrane potential collapse in the human dermal fibroblast primary cells. Iran. J. Pharm. Res. 2012, 11, 495–501.
- Shaki, F.; Pourahmad, J. Mitochondrial toxicity of depleted uranium: Protection by Beta-glucan. Iran. J. Pharm. Res. 2013, 12, 131–140.
- Sun, M.H.; Liu, S.Q.; Du, K.J.; Nie, C.M.; Lin, Y.W. A spectroscopic study of uranyl-cytochrome b5/cytochrome c interactions. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2014, 118, 130–137.
- Thiébault, C.; Carriere, M.; Milgram, S.; Simon, A.; Avoscan, L.; Gouget, B. Uranium induces apoptosis and is genotoxic to normal rat kidney (NRK-52E) proximal cells. Toxicol. Sci. 2007, 98, 479–487.
- Liu, F.; Du, K.J.; Fang, Z.; You, Y.; Wen, G.B.; Lin, Y.W. Chemical and biological insights into uranium-induced apoptosis of rat hepatic cell line. Radiat. Environ. Biophys. 2015, 54, 207–216.
- Hao, Y.; Ren, J.; Liu, C.; Li, H.; Liu, J.; Yang, Z.; Li, R.; Su, Y. Zinc protects human kidney cells from depleted uranium-induced apoptosis. Basic Clin. Pharmacol. Toxicol. 2013, 114, 271–280.
- Burbank, K.A.; Walker, R.A.; Peyton, B.M. A molecular level mechanism for uranium (VI) toxicity through Ca2+ displacement in pyrroloquinoline quinone-dependent bacterial dehydrogenase. J. Inorg. Biochem. 2015, 149, 59–67.
- Tipping, E.; Lofts, S. Testing WHAM-FTOX with laboratory toxicity data for mixtures of metals (Cu, Zn, Cd, Ag, Pb). Environ. Toxicol. Chem. 2015, 34, 788–798.
- Khan, M.J.; Mukhtiar, M.; Qureshi, M.M.; Jan, S.U.; Ullah, I.; Hussain, A.; Khan, M.F.; Gul, R.; Shahwani, N.A.; Rabbani, I. Spectrophotometric investigation of glutathione modulation by thallium chloride in aqueous medium. Pak. J. Pharm. Sci. 2018, 31, 1463–1467.
- Miranda, R.R.; Bezerra, A.G., Jr.; Oliveira Ribeiro, C.A.; Randi, M.A.; Voigt, C.L.; Skytte, L.; Rasmussen, K.L.; Kjeldsen, F.; Filipak Neto, F. Toxicological interactions of silver nanoparticles and non-essential metals in human hepatocarcinoma cell line. Toxicol. In Vitro 2017, 40, 134–143.
- Guan, S.; Tao, S.; Huang, Y.; Jin, Y.; Hu, Y.; Lu, J. Combined toxic effects of CBNPs and Pb on rat alveolar macrophage apoptosis and autophagy flux. Ecotoxicol. Environ. Saf. 2020, 205, 111062.
- Fiocchetti, M.; Cipolletti, M.; Leone, S.; Naldini, A.; Carraro, F.; Giordano, D.; Verde, C.; Ascenzi, P.; Marino, M. Neuroglobin in breast cancer cells: Effect of hypoxia and oxidative stress on protein level, localization, and anti-apoptotic function. PLoS ONE 2016, 11, e0154959.
- Bragadin, M.; Marton, D.; Murgia, M.; Rizzoli, V.; Scutari, G.; Deana, R. Interactions of trialkyllead compounds with rat liver mitochondria. J. Inorg. Biochem. 1998, 69, 259–262.
- Aldridge, W.N.; Street, B.W.; Skilleter, D.N. Oxidative phosphorylation. Halide-dependent and halide-independent effects of triorganotin and trioganolead compounds on mitochondrial functions. Biochem. J. 1977, 168, 353–364.
- Skilleter, D.N. The decrease of mitochondrial substrate uptake caused by trialkyltin and trialkyl-lead compounds in chloride media and its relevance to inhibition of oxidative phosphorylation. Biochem. J. 1975, 146, 465–4671.
- Kapetana, M.; Banti, C.N.; Papachristodoulou, C.; Psycharis, V.; Raptopoulou, C.P.; Hadjikakou, S.K. Conjugation of triphenylantimony(V) with carvacrol against human breast cancer cells. J. Biol. Inorg. Chem. 2022, 27, 373–389.
- Issartel, J.P.; Dupuis, A.; Lunardi, J.; Vignais, P.V. Fluoroaluminum and fluoroberyllium nucleoside diphosphate complexes as probes of the enzymatic mechanism of the mitochondrial F1-ATPase. Biochemistry 1991, 30, 4726–4733.
- Missiaen, L.; Wuytack, F.; De Smedt, H.; Vrolix, M.; Casteels, R. AlF4− reversibly inhibits ‘P’-type cation-transport ATPases, possibly by interacting with the phosphate-binding site of the ATPase. Biochem. J. 1988, 253, 827–833.
- Braig, K.; Menz, R.I.; Montgomery, M.G.; Leslie, A.G.; Walker, J.E. Structure of bovine mitochondrial F1-ATPase inhibited by Mg2+ ADP and aluminium fluoride. Structure 2000, 8, 567–573.
- Tunes, L.G.; Morato, R.E.; Garcia, A.; Schmitz, V.; Steindel, M.; Corrêa-Junior, J.D.; Dos Santos, H.F.; Frézard, F.; de Almeida, M.V.; Silva, H.; et al. Preclinical gold complexes as oral drug candidates to treat leishmaniasis are potent trypanothione reductase inhibitors. ACS Infect. Dis. 2020, 6, 1121–1139.
This entry is offline, you can click here to edit this entry!