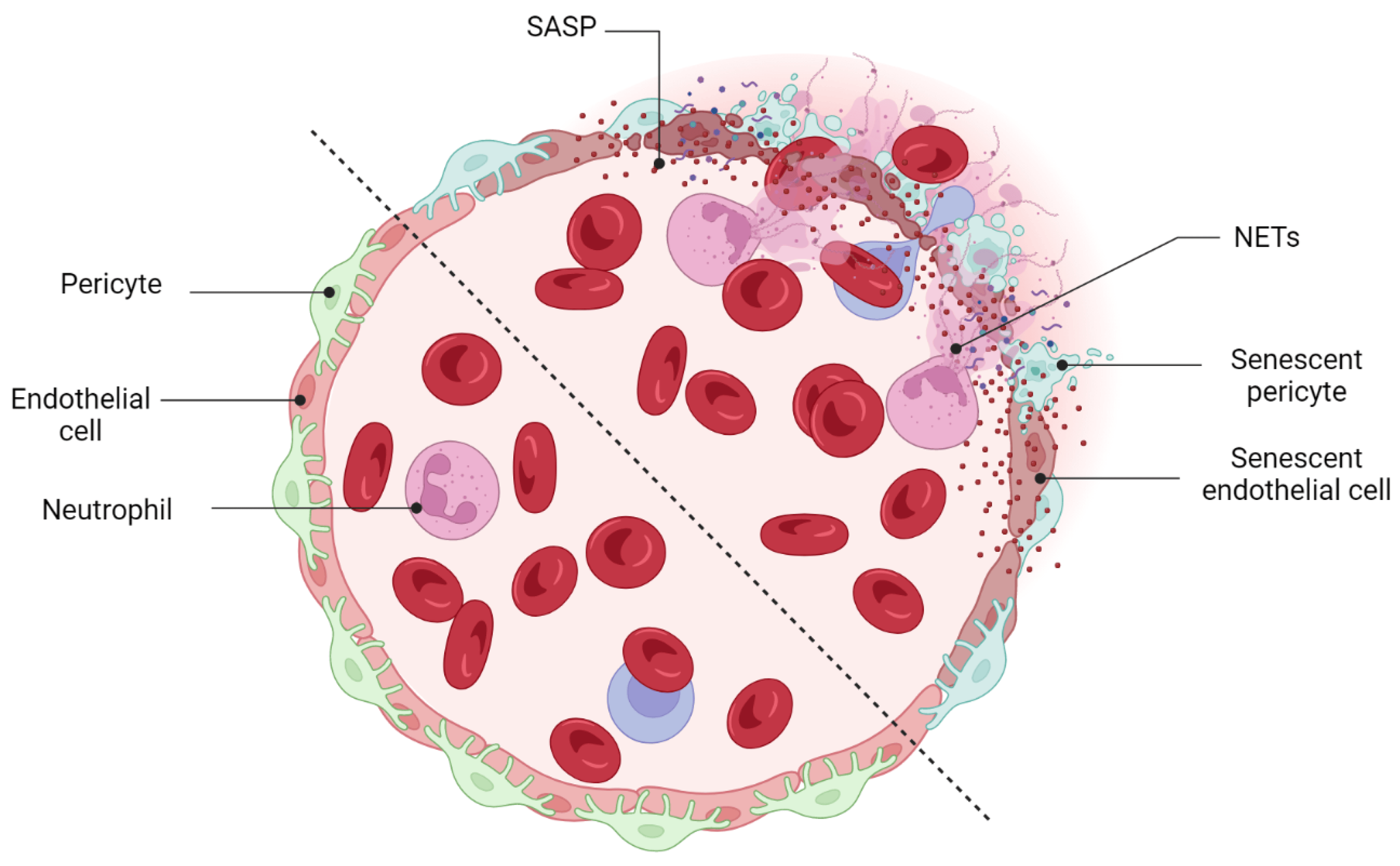
Cellular senescence, a state of permanent cell cycle arrest in response to endogenous and exogenous stimuli, triggers a series of gradual alterations in structure, metabolism, and function, as well as inflammatory gene expression that nurtures a low-grade proinflammatory milieu in human tissue. A growing body of evidence indicates an accumulation of senescent neurons and blood vessels in response to stress and aging in the retina. Prolonged accumulation of senescent cells and long-term activation of stress signaling responses may lead to multiple chronic diseases, tissue dysfunction, and age-related pathologies by exposing neighboring cells to the heightened pathological senescence-associated secretory phenotype (SASP).
- cellular senescence
- senescence-associated secretory phenotype (SASP)
- retinal vascular system
1. Cellular Senescence
1.1. Cell Senescence and Inducers
1.2. Cell Cycle Arrest
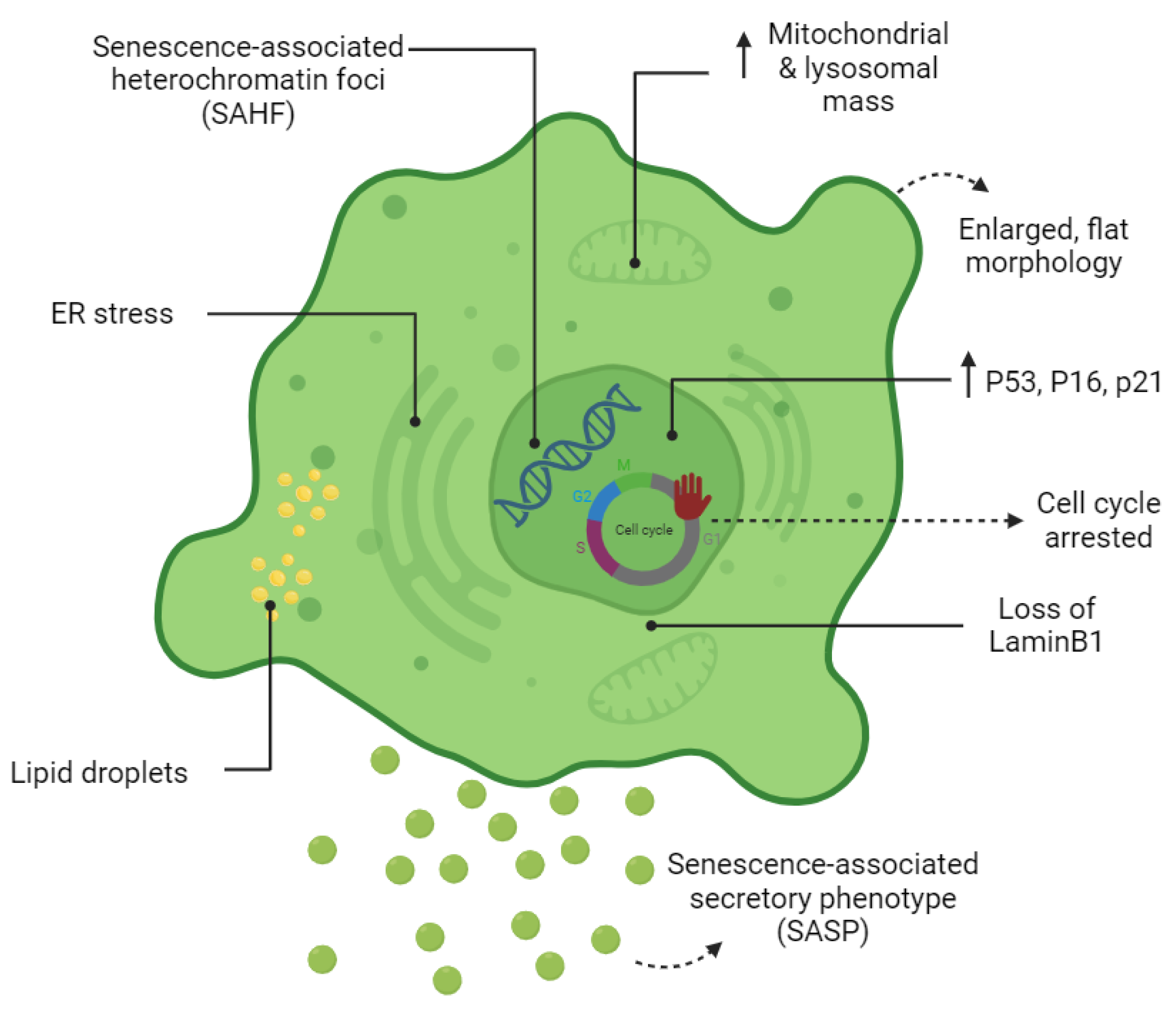
1.3. The Senescent Phenotype
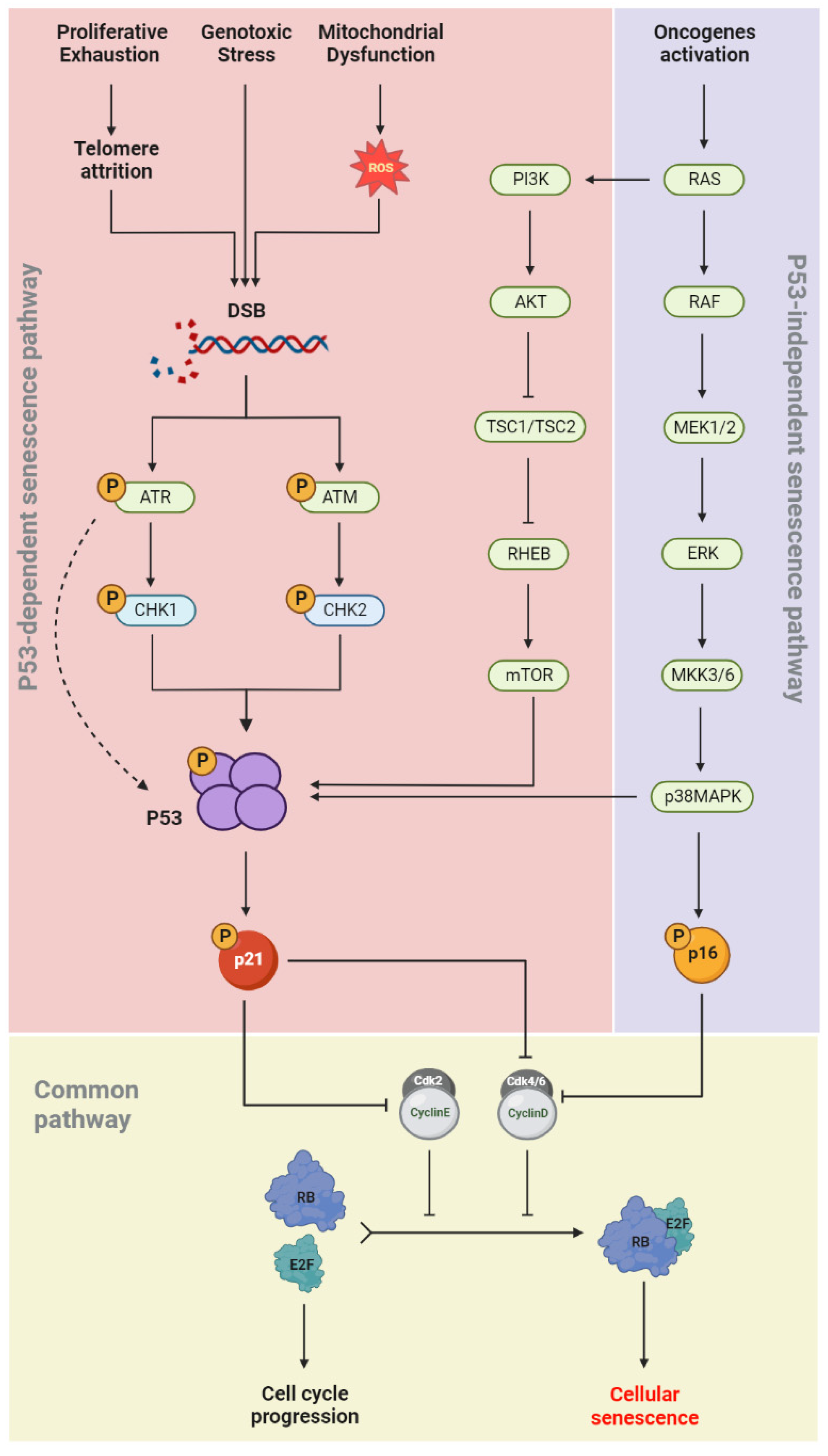
1.4. Senescence Molecular Pathways
1.5. Autophagy: A Pro-Senescence or an Anti-Senescence Mechanism?
2. Implications of SCs on the Retinal Vascular System
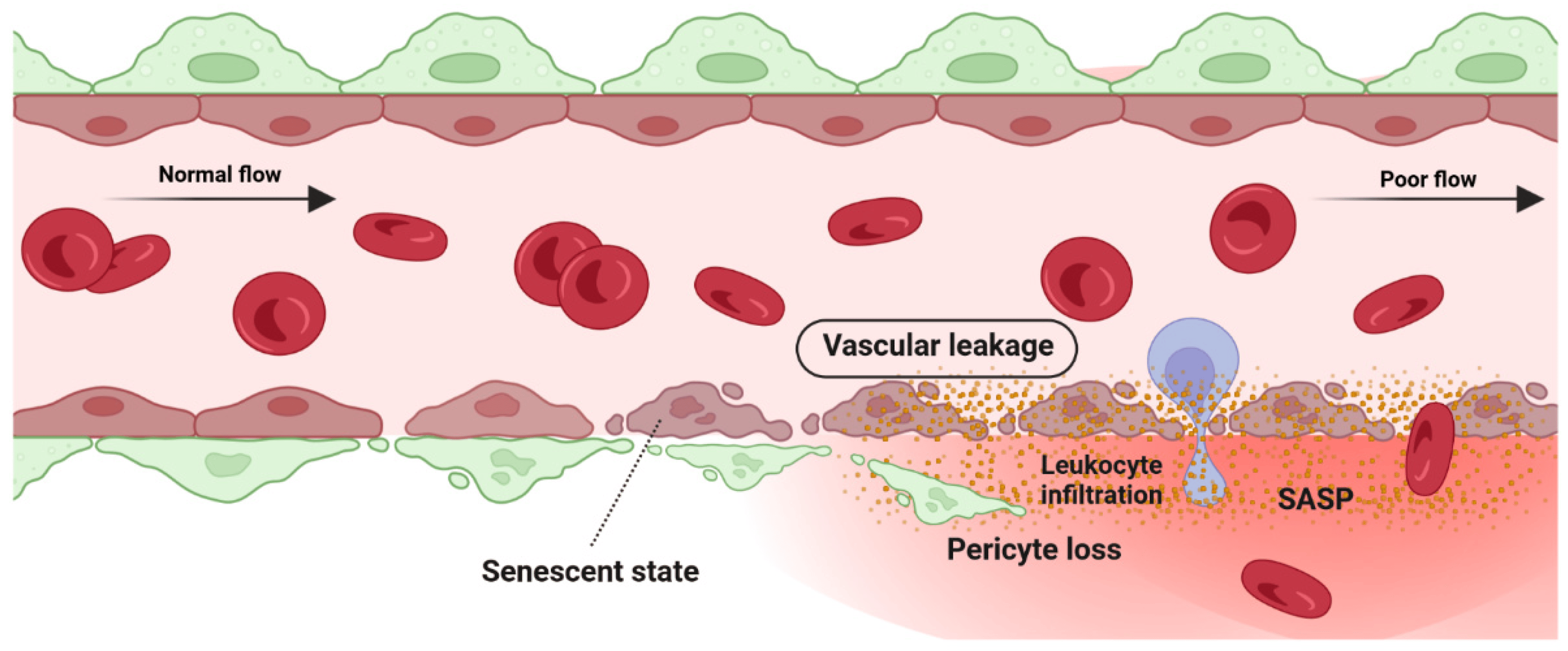
2.1. Detrimental Effect: Vascular Degeneration
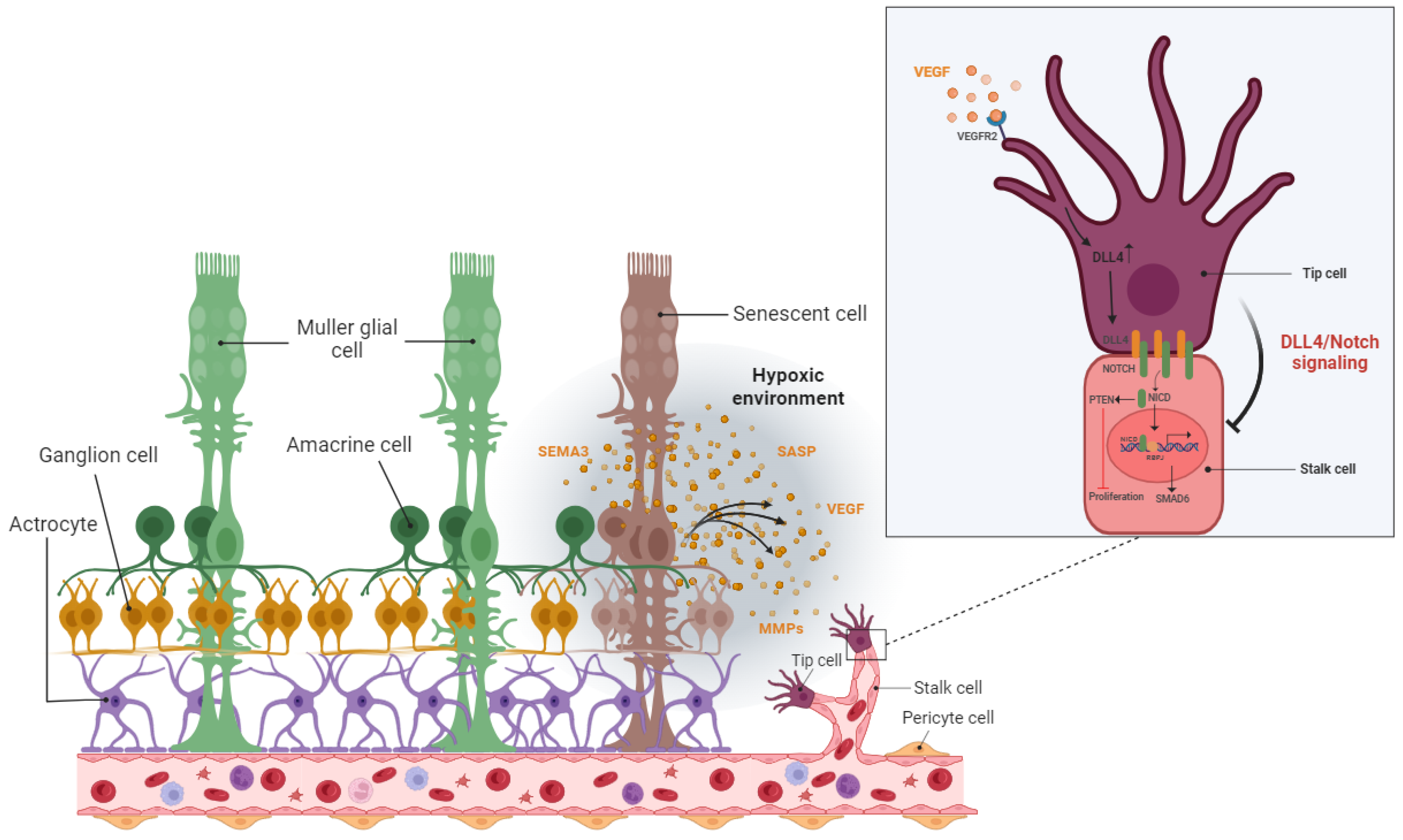
2.2. Detrimental Effect: Pathological Angiogenesis
2.3. Beneficial Effect: Vascular Remodeling
This entry is adapted from the peer-reviewed paper 10.3390/cells12192341
References
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593.
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621.
- Vasileiou, P.V.S.; Evangelou, K.; Vlasis, K.; Fildisis, G.; Panayiotidis, M.I.; Chronopoulos, E.; Passias, P.G.; Kouloukoussa, M.; Gorgoulis, V.G.; Havaki, S. Mitochondrial Homeostasis and Cellular Senescence. Cells 2019, 8, 686.
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 2022, 23, 74–88.
- Zhao, M.L.; Rabiee, A.; Kovary, K.M.; Bahrami-Nejad, Z.; Taylor, B.; Teruel, M.N. Molecular Competition in G1 Controls When Cells Simultaneously Commit to Terminally Differentiate and Exit the Cell Cycle. Cell Rep. 2020, 31, 107769.
- Ruijtenberg, S.; van den Heuvel, S. Coordinating cell proliferation and differentiation: Antagonism between cell cycle regulators and cell type-specific gene expression. Cell Cycle 2016, 15, 196–212.
- Rumman, M.; Dhawan, J.; Kassem, M. Concise Review: Quiescence in Adult Stem Cells: Biological Significance and Relevance to Tissue Regeneration. Stem. Cells 2015, 33, 2903–2912.
- Maciel-Baron, L.A.; Moreno-Blas, D.; Morales-Rosales, S.L.; Gonzalez-Puertos, V.Y.; Lopez-Diazguerrero, N.E.; Torres, C.; Castro-Obregon, S.; Konigsberg, M. Cellular Senescence, Neurological Function, and Redox State. Antioxid Redox. Signal 2018, 28, 1704–1723.
- Cho, I.J.; Lui, P.P.; Obajdin, J.; Riccio, F.; Stroukov, W.; Willis, T.L.; Spagnoli, F.; Watt, F.M. Mechanisms, Hallmarks, and Implications of Stem Cell Quiescence. Stem Cell Rep. 2019, 12, 1190–1200.
- De Morree, A.; Rando, T.A. Regulation of adult stem cell quiescence and its functions in the maintenance of tissue integrity. Nat. Rev. Mol. Cell Biol. 2023, 24, 334–354.
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114.
- Vergel, M.; Marin, J.J.; Estevez, P.; Carnero, A. Cellular senescence as a target in cancer control. J. Aging Res. 2010, 2011, 725365.
- Leikam, C.; Hufnagel, A.; Schartl, M.; Meierjohann, S. Oncogene activation in melanocytes links reactive oxygen to multinucleated phenotype and senescence. Oncogene 2008, 27, 7070–7082.
- Takahashi, A.; Ohtani, N.; Yamakoshi, K.; Iida, S.; Tahara, H.; Nakayama, K.; Nakayama, K.I.; Ide, T.; Saya, H.; Hara, E. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat. Cell Biol. 2006, 8, 1291–1297.
- Panopoulos, A.; Pacios-Bras, C.; Choi, J.; Yenjerla, M.; Sussman, M.A.; Fotedar, R.; Margolis, R.L. Failure of cell cleavage induces senescence in tetraploid primary cells. Mol. Biol. Cell 2014, 25, 3105–3118.
- Walen, K.H. Human diploid fibroblast cells in senescence; cycling through polyploidy to mitotic cells. Vitr. Cell Dev. Biol. Anim. 2006, 42, 216–224.
- Dikovskaya, D.; Cole, J.J.; Mason, S.M.; Nixon, C.; Karim, S.A.; McGarry, L.; Clark, W.; Hewitt, R.N.; Sammons, M.A.; Zhu, J.; et al. Mitotic Stress Is an Integral Part of the Oncogene-Induced Senescence Program that Promotes Multinucleation and Cell Cycle Arrest. Cell Rep. 2015, 12, 1483–1496.
- Rovira, M.; Sereda, R.; Pladevall-Morera, D.; Ramponi, V.; Marin, I.; Maus, M.; Madrigal-Matute, J.; Diaz, A.; Garcia, F.; Munoz, J.; et al. The lysosomal proteome of senescent cells contributes to the senescence secretome. Aging Cell 2022, 21, e13707.
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367.
- Severino, J.; Allen, R.G.; Balin, S.; Balin, A.; Cristofalo, V.J. Is beta-galactosidase staining a marker of senescence in vitro and in vivo? Exp. Cell Res. 2000, 257, 162–171.
- Terman, A.; Brunk, U.T. The aging myocardium: Roles of mitochondrial damage and lysosomal degradation. Heart Lung Circ. 2005, 14, 107–114.
- Brunk, U.T.; Terman, A. Lipofuscin: Mechanisms of age-related accumulation and influence on cell function. Free Radic Biol. Med. 2002, 33, 611–619.
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408.
- Saraste, M. Oxidative phosphorylation at the fin de siecle. Science 1999, 283, 1488–1493.
- James, E.L.; Michalek, R.D.; Pitiyage, G.N.; de Castro, A.M.; Vignola, K.S.; Jones, J.; Mohney, R.P.; Karoly, E.D.; Prime, S.S.; Parkinson, E.K. Senescent human fibroblasts show increased glycolysis and redox homeostasis with extracellular metabolomes that overlap with those of irreparable DNA damage, aging, and disease. J. Proteome Res. 2015, 14, 1854–1871.
- Parkinson, E.K.; Adamski, J.; Zahn, G.; Gaumann, A.; Flores-Borja, F.; Ziegler, C.; Mycielska, M.E. Extracellular citrate and metabolic adaptations of cancer cells. Cancer Metastasis Rev. 2021, 40, 1073–1091.
- Zwerschke, W.; Mazurek, S.; Stockl, P.; Hutter, E.; Eigenbrodt, E.; Jansen-Durr, P. Metabolic analysis of senescent human fibroblasts reveals a role for AMP in cellular senescence. Biochem. J. 2003, 376, 403–411.
- Miwa, S.; Kashyap, S.; Chini, E.; von Zglinicki, T. Mitochondrial dysfunction in cell senescence and aging. J. Clin. Invest. 2022, 132, e158447.
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007, 5, e110.
- Petersen, S.; Saretzki, G.; von Zglinicki, T. Preferential accumulation of single-stranded regions in telomeres of human fibroblasts. Exp. Cell Res. 1998, 239, 152–160.
- Manzella, N.; Santin, Y.; Maggiorani, D.; Martini, H.; Douin-Echinard, V.; Passos, J.F.; Lezoualc’h, F.; Binda, C.; Parini, A.; Mialet-Perez, J. Monoamine oxidase-A is a novel driver of stress-induced premature senescence through inhibition of parkin-mediated mitophagy. Aging Cell 2018, 17, e12811.
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314.
- Roger, L.; Tomas, F.; Gire, V. Mechanisms and Regulation of Cellular Senescence. Int. J. Mol. Sci. 2021, 22, 13173.
- Cormenier, J.; Martin, N.; Desle, J.; Salazar-Cardozo, C.; Pourtier, A.; Abbadie, C.; Pluquet, O. The ATF6alpha arm of the Unfolded Protein Response mediates replicative senescence in human fibroblasts through a COX2/prostaglandin E2 intracrine pathway. Mech. Ageing Dev. 2018, 170, 82–91.
- Ohno-Iwashita, Y.; Shimada, Y.; Hayashi, M.; Inomata, M. Plasma membrane microdomains in aging and disease. Geriatr. Gerontol. Int. 2010, 10 (Suppl 1), S41–S52.
- Narita, M.; Nunez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716.
- Di Micco, R.; Sulli, G.; Dobreva, M.; Liontos, M.; Botrugno, O.A.; Gargiulo, G.; dal Zuffo, R.; Matti, V.; d’Ario, G.; Montani, E.; et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell Biol. 2011, 13, 292–302.
- d’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198.
- Reinhardt, H.C.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012, 28, 128–136.
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112.
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138.
- Herbig, U.; Jobling, W.A.; Chen, B.P.; Chen, D.J.; Sedivy, J.M. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol. Cell 2004, 14, 501–513.
- Serrano, M.; Hannon, G.J.; Beach, D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993, 366, 704–707.
- Young, A.R.J.; Cassidy, L.D.; Narita, M. Autophagy and senescence, converging roles in pathophysiology as seen through mouse models. Adv. Cancer Res. 2021, 150, 113–145.
- Kwon, Y.; Kim, J.W.; Jeoung, J.A.; Kim, M.S.; Kang, C. Autophagy is pro-senescence when seen in close-up, but anti-senescence in long-shot. Mol. Cells 2017, 40, 607–612.
- Garcia-Prat, L.; Martinez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodriguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42.
- Wen, X.; Klionsky, D.J. Autophagy is a key factor in maintaining the regenerative capacity of muscle stem cells by promoting quiescence and preventing senescence. Autophagy 2016, 12, 1158373.
- Kang, H.T.; Lee, K.B.; Kim, S.Y.; Choi, H.R.; Park, S.C. Autophagy impairment induces premature senescence in primary human fibroblasts. PLoS ONE 2011, 6, e23367.
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavare, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803.
- Narita, M.; Young, A.R.; Arakawa, S.; Samarajiwa, S.A.; Nakashima, T.; Yoshida, S.; Hong, S.; Berry, L.S.; Reichelt, S.; Ferreira, M.; et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 2011, 332, 966–970.
- Nam, H.Y.; Han, M.W.; Chang, H.W.; Kim, S.Y.; Kim, S.W. Prolonged autophagy by MTOR inhibitor leads radioresistant cancer cells into senescence. Autophagy 2013, 9, 1631–1632.
- Liu, H.; Prokosch, V. Energy Metabolism in the Inner Retina in Health and Glaucoma. Int. J. Mol. Sci. 2021, 22, 3689.
- Oubaha, M.; Miloudi, K.; Dejda, A.; Guber, V.; Mawambo, G.; Germain, M.A.; Bourdel, G.; Popovic, N.; Rezende, F.A.; Kaufman, R.J.; et al. Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci. Transl. Med. 2016, 8, 362ra144.
- Sapieha, P.; Mallette, F.A. Cellular Senescence in Postmitotic Cells: Beyond Growth Arrest. Trends Cell Biol. 2018, 28, 595–607.
- Brown, B.A.; Williams, H.; George, S.J. Evidence for the Involvement of Matrix-Degrading Metalloproteinases (MMPs) in Atherosclerosis. Prog. Mol. Biol. Transl. Sci. 2017, 147, 197–237.
- Bertelli, P.M.; Pedrini, E.; Hughes, D.; McDonnell, S.; Pathak, V.; Peixoto, E.; Guduric-Fuchs, J.; Stitt, A.W.; Medina, R.J. Long term high glucose exposure induces premature senescence in retinal endothelial cells. Front. Physiol. 2022, 13, 929118.
- Tian, X.L.; Li, Y. Endothelial cell senescence and age-related vascular diseases. J. Genet. Genom. 2014, 41, 485–495.
- Mylonas, A.; O’Loghlen, A. Cellular Senescence and Ageing: Mechanisms and Interventions. Front. Aging 2022, 3, 866718.
- Del Cuore, A.; Pacinella, G.; Riolo, R.; Tuttolomondo, A. The Role of Immunosenescence in Cerebral Small Vessel Disease: A Review. Int. J. Mol. Sci. 2022, 23, 7136.
- Cheung, T.M.; Ganatra, M.P.; Peters, E.B.; Truskey, G.A. Effect of cellular senescence on the albumin permeability of blood-derived endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H1374–H1383.
- Yamazaki, Y.; Baker, D.J.; Tachibana, M.; Liu, C.C.; van Deursen, J.M.; Brott, T.G.; Bu, G.; Kanekiyo, T. Vascular Cell Senescence Contributes to Blood-Brain Barrier Breakdown. Stroke 2016, 47, 1068–1077.
- Venkatesh, D.; Fredette, N.; Rostama, B.; Tang, Y.; Vary, C.P.; Liaw, L.; Urs, S. RhoA-mediated signaling in Notch-induced senescence-like growth arrest and endothelial barrier dysfunction. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 876–882.
- Krouwer, V.J.; Hekking, L.H.; Langelaar-Makkinje, M.; Regan-Klapisz, E.; Post, J.A. Endothelial cell senescence is associated with disrupted cell-cell junctions and increased monolayer permeability. Vasc. Cell 2012, 4, 12.
- Wajapeyee, N.; Serra, R.W.; Zhu, X.; Mahalingam, M.; Green, M.R. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell 2008, 132, 363–374.
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031.
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018.
- Crespo-Garcia, S.; Tsuruda, P.R.; Dejda, A.; Ryan, R.D.; Fournier, F.; Chaney, S.Y.; Pilon, F.; Dogan, T.; Cagnone, G.; Patel, P.; et al. Pathological angiogenesis in retinopathy engages cellular senescence and is amenable to therapeutic elimination via BCL-xL inhibition. Cell Metab. 2021, 33, 818–832.e817.
- Yoshikawa, Y.; Yamada, T.; Tai-Nagara, I.; Okabe, K.; Kitagawa, Y.; Ema, M.; Kubota, Y. Developmental regression of hyaloid vasculature is triggered by neurons. J. Exp. Med. 2016, 213, 1175–1183.
- Hamrick, S.E.G.; Sallmon, H.; Rose, A.T.; Porras, D.; Shelton, E.L.; Reese, J.; Hansmann, G. Patent Ductus Arteriosus of the Preterm Infant. Pediatrics 2020, 146, e20201209.
- Girling, J.E.; Rogers, P.A. Regulation of endometrial vascular remodelling: Role of the vascular endothelial growth factor family and the angiopoietin-TIE signalling system. Reproduction 2009, 138, 883–893.
- Binet, F.; Cagnone, G.; Crespo-Garcia, S.; Hata, M.; Neault, M.; Dejda, A.; Wilson, A.M.; Buscarlet, M.; Mawambo, G.T.; Howard, J.P.; et al. Neutrophil extracellular traps target senescent vasculature for tissue remodeling in retinopathy. Science 2020, 369, eaay5356.
- Hartnett, M.E.; Penn, J.S. Mechanisms and management of retinopathy of prematurity. N. Engl. J. Med. 2012, 367, 2515–2526.
- Klotzsche-von Ameln, A.; Sprott, D. Harnessing retinal phagocytes to combat pathological neovascularization in ischemic retinopathies? Pflug. Arch. 2022, 474, 575–590.
- Ju, R.H.; Zhang, J.Q.; Ke, X.Y.; Lu, X.H.; Liang, L.F.; Wang, W.J. Spontaneous regression of retinopathy of prematurity: Incidence and predictive factors. Int. J. Ophthalmol. 2013, 6, 475–480.
- Han, J.R.; Ju, W.K.; Park, I.W. Spontaneous regression of neovascularization at the disc in diabetic retinopathy. Korean J. Ophthalmol. 2004, 18, 41–46.
- Bandello, F.; Gass, J.D.; Lattanzio, R.; Brancato, R. Spontaneous regression of neovascularization at the disk and elsewhere in diabetic retinopathy. Am. J. Ophthalmol. 1996, 122, 494–501.
- Gao, Y.; Galis, Z.S. Exploring the Role of Endothelial Cell Resilience in Cardiovascular Health and Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 179–185.
- Melo, L.G.; Gnecchi, M.; Ward, C.A.; Dzau, V.J. Vascular Remodeling in Health and Disease. J. Cardiovasc. Med. 2007, 1541–1565.
- Eelen, G.; de Zeeuw, P.; Treps, L.; Harjes, U.; Wong, B.W.; Carmeliet, P. Endothelial Cell Metabolism. Physiol. Rev. 2018, 98, 3–58.
- Egashira, M.; Hirota, Y.; Shimizu-Hirota, R.; Saito-Fujita, T.; Haraguchi, H.; Matsumoto, L.; Matsuo, M.; Hiraoka, T.; Tanaka, T.; Akaeda, S.; et al. F4/80+ Macrophages Contribute to Clearance of Senescent Cells in the Mouse Postpartum Uterus. Endocrinology 2017, 158, 2344–2353.
- Nacher, V.; Carretero, A.; Navarro, M.; Armengol, C.; Llombart, C.; Rodriguez, A.; Herrero-Fresneda, I.; Ayuso, E.; Ruberte, J. The quail mesonephros: A new model for renal senescence? J. Vasc. Res. 2006, 43, 581–586.
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551.
- Deliyanti, D.; Talia, D.M.; Zhu, T.; Maxwell, M.J.; Agrotis, A.; Jerome, J.R.; Hargreaves, E.M.; Gerondakis, S.; Hibbs, M.L.; Mackay, F.; et al. Foxp3(+) Tregs are recruited to the retina to repair pathological angiogenesis. Nat. Commun. 2017, 8, 748.
- Simo, R.; Hernandez, C. Novel approaches for treating diabetic retinopathy based on recent pathogenic evidence. Prog. Retin. Eye Res. 2015, 48, 160–180.
- Agarwal, A.; Rhoades, W.R.; Hanout, M.; Soliman, M.K.; Sarwar, S.; Sadiq, M.A.; Sepah, Y.J.; Do, D.V.; Nguyen, Q.D. Management of neovascular age-related macular degeneration: Current state-of-the-art care for optimizing visual outcomes and therapies in development. Clin. Ophthalmol. 2015, 9, 1001–1015.
- Sapieha, P.; Joyal, J.S.; Rivera, J.C.; Kermorvant-Duchemin, E.; Sennlaub, F.; Hardy, P.; Lachapelle, P.; Chemtob, S. Retinopathy of prematurity: Understanding ischemic retinal vasculopathies at an extreme of life. J. Clin. Invest. 2010, 120, 3022–3032.
- Altmann, C.; Schmidt, M.H.H. The Role of Microglia in Diabetic Retinopathy: Inflammation, Microvasculature Defects and Neurodegeneration. Int. J. Mol. Sci. 2018, 19, 110.
- Semeraro, F.; Cancarini, A.; dell’Omo, R.; Rezzola, S.; Romano, M.R.; Costagliola, C. Diabetic Retinopathy: Vascular and Inflammatory Disease. J. Diabetes Res. 2015, 2015, 582060.
- Arroba, A.I.; Alcalde-Estevez, E.; Garcia-Ramirez, M.; Cazzoni, D.; de la Villa, P.; Sanchez-Fernandez, E.M.; Mellet, C.O.; Garcia Fernandez, J.M.; Hernandez, C.; Simo, R.; et al. Modulation of microglia polarization dynamics during diabetic retinopathy in db/db mice. Biochim. Biophys. Acta 2016, 1862, 1663–1674.
- Zeng, H.Y.; Green, W.R.; Tso, M.O. Microglial activation in human diabetic retinopathy. Arch. Ophthalmol. 2008, 126, 227–232.
- Li, J.; Yu, S.; Lu, X.; Cui, K.; Tang, X.; Xu, Y.; Liang, X. The phase changes of M1/M2 phenotype of microglia/macrophage following oxygen-induced retinopathy in mice. Inflamm. Res. 2021, 70, 183–192.
- Zhou, Y.; Yoshida, S.; Nakao, S.; Yoshimura, T.; Kobayashi, Y.; Nakama, T.; Kubo, Y.; Miyawaki, K.; Yamaguchi, M.; Ishikawa, K.; et al. M2 Macrophages Enhance Pathological Neovascularization in the Mouse Model of Oxygen-Induced Retinopathy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 4767–4777.
- Korovina, I.; Neuwirth, A.; Sprott, D.; Troullinaki, M.; Poitz, D.M.; Deussen, A.; Klotzsche-von Ameln, A. Myeloid SOCS3 Deficiency Regulates Angiogenesis via Enhanced Apoptotic Endothelial Cell Engulfment. J. Innate Immun. 2020, 12, 248–256.
- Korovina, I.; Neuwirth, A.; Sprott, D.; Weber, S.; Sardar Pasha, S.P.B.; Gercken, B.; Breier, G.; El-Armouche, A.; Deussen, A.; Karl, M.O.; et al. Hematopoietic hypoxia-inducible factor 2alpha deficiency ameliorates pathological retinal neovascularization via modulation of endothelial cell apoptosis. FASEB J. 2019, 33, 1758–1770.
- He, P. Leucocyte/endothelium interactions and microvessel permeability: Coupled or uncoupled? Cardiovasc. Res. 2010, 87, 281–290.
- Joussen, A.M.; Poulaki, V.; Mitsiades, N.; Cai, W.Y.; Suzuma, I.; Pak, J.; Ju, S.T.; Rook, S.L.; Esser, P.; Mitsiades, C.S.; et al. Suppression of Fas-FasL-induced endothelial cell apoptosis prevents diabetic blood-retinal barrier breakdown in a model of streptozotocin-induced diabetes. FASEB J. 2003, 17, 76–78.
- Davies, M.H.; Stempel, A.J.; Powers, M.R. MCP-1 deficiency delays regression of pathologic retinal neovascularization in a model of ischemic retinopathy. Invest. Ophthalmol. Vis. Sci. 2008, 49, 4195–4202.
- Wang, L.; Zhou, X.; Yin, Y.; Mai, Y.; Wang, D.; Zhang, X. Hyperglycemia Induces Neutrophil Extracellular Traps Formation Through an NADPH Oxidase-Dependent Pathway in Diabetic Retinopathy. Front. Immunol. 2018, 9, 3076.
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819.
- Menegazzo, L.; Ciciliot, S.; Poncina, N.; Mazzucato, M.; Persano, M.; Bonora, B.; Albiero, M.; Vigili de Kreutzenberg, S.; Avogaro, A.; Fadini, G.P. NETosis is induced by high glucose and associated with type 2 diabetes. Acta Diabetol. 2015, 52, 497–503.
- Park, J.H.; Kim, J.E.; Gu, J.Y.; Yoo, H.J.; Park, S.H.; Kim, Y.I.; Nam-Goong, I.S.; Kim, E.S.; Kim, H.K. Evaluation of Circulating Markers of Neutrophil Extracellular Trap (NET) Formation as Risk Factors for Diabetic Retinopathy in a Case-Control Association Study. Exp. Clin. Endocrinol. Diabetes 2016, 124, 557–561.