Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Infectious Diseases
Sickle cell disease (SCD) is a complex genetic disorder associated with multiple clinical manifestations, including increased susceptibility to bacterial and viral infections.
- sickle cell disease
- infection
- bacteria
- virus
- molecular mechanism
1. Introduction
Sickle cell disease (SCD) is caused by a single amino acid substitution (Glu > Val) at codon six at the globin gene, which results in variant hemoglobin formation (HbSS). This causes an increase in viscosity and adhesion to vascular walls, resulting in obstructing the blood flow in small capillaries [1]. This primary pathophysiological condition has several clinical manifestations, such as vaso-occlusive crisis (VOC), splenomegaly, acute chest syndrome (ACS), ocular manifestation, hepatomegaly, pulmonary hypertension, leg ulcers, chronic kidney disease (CKD), and stroke, which leads to early mortality [2][3][4][5]. However, in the current scenario, hydroxyurea (HU) is the only SCD drug approved by the Food and Drug Administration (FDA) to increase fetal hemoglobin (HbF) levels [6].
The phenotypic manifestation of the disease is still poorly understood. However, environmental factors, including climate and air quality, infections, fetal hemoglobin levels, and other genetic factors, play a vital role in the progression of the disease [7][8]. The severity of the disease in pediatric SCD patients, particularly those under the age of five years, is increased for various reasons, including rapid sequestration of red blood cells in the spleen, failure of opsonization, and an inability to deal with infective encapsulated microorganisms after infection [9][10]. As a result, in children with SCD, infection is the second leading cause of death in their first ten years.
SCD patients in sub-Saharan Africa and the Eastern Mediterranean have a high infection rate, with obtrusive pneumococcal infection being the most common [11]. The global reports suggest implementing an effective management and systemic newborn screening (NBS) program is the first logical step in preventing disease [11]. The initial neonatal screening and the arrangement of immunization and prophylactic antimicrobial agents improve the quality of care for SCD patients, resulting in a significant decrease in mortality.
2. Infection of Childhood Mortality in Sickle Cell Disease
2.1. Immune Dysfunction and Susceptibility to Infection
In patients with SCD, the primary complications often arise from infections. The exact reason behind susceptibility to infections is complicated. A significant contributing factor is the impaired functioning of the spleen. SCD has been associated with abnormalities in processes such as opsonization, the alternate complement pathway, antibody production, leucocyte function, and cell-mediated immunity.
Individuals with SCD exhibit deficiencies in their innate immune system, primarily attributed to reduced splenic function, making them more susceptible to infections caused by encapsulated bacteria, and the compromised innate immune response stems from malfunctions in elements like chemotaxis, migration, and scavenging due to faulty neutrophils, alongside decreased splenic phagocytic filtration and a diminished opsonization capability [10].
2.2. Splenic Dysfunction
The spleen is primarily involved in the filtration of foreign microorganisms and the support of innate and adaptive immune functions. It serves various vital functions in immune response and plays a critical role in increasing vulnerability to most SCD patients’ infections. The spleen helps two essential tasks prevent bloodstream bacterial infections. First, it is a phagocytic filter that removes bacteria from the bloodstream. Second, the spleen hosts leucocytes, which produce an antibody response to polysaccharide antigens [12]. Still, most infections are caused by SCD complications, such as acidosis, hypoxia, and dehydration, which activate or exaggerate the sickle cell crises and cause vaso-occlusion and ischemia, ultimately harming the structure and function of the spleen [13].
Individuals with splenectomy and individuals with non- or partially functioning spleens (hypo or asplenism), as in SCD, are more vulnerable to infections. Hyposplenic and asplenic individuals lack IgM memory B cells (as stated above, the spleen is a major site of their generation or function). Thus, it cannot provide a rapid, precise response to encapsulated species of microorganisms, particularly Haemophilus influenzae type b (Hib), salmonellae, and pneumococcal infections [14]. Local infections can easily become systemic, which, combined with the loss of spleen filtering function, can contribute to the development of severe sepsis. The risk of infection in children under the age of five with SCD is nearly thirty to a hundred times higher than in healthy children, implying that SCD children are more vulnerable to invasive pneumococcal disease (IPD), including pneumonia, meningitis, and septicemia [15].
2.3. Opsonization
Splenic opsonization is compromised in SCD conditions due to a lack of production of the immunoglobins and opsonins required for bacterial destruction. The effective opsonization process is dependent on a complement cascade, which is activated by both classical and alternative pathways and destroys infectious microorganisms by creating holes in their cell membranes [16]. Lack of splenic IgM leads to decreased opsonization and impaired activation of the classical pathway. Another opsonin, tuftsin, formed in the spleen and required to activate cell-mediated immunity, is reduced in SCD patients, indicating that the spleen is involved in their synthesis [17].
2.4. Lymphocytes
SCD patients are more susceptible to unusual pathogens, implying that other immune system defects compromise their immune systems. SCD reduces the production and function of B and T lymphocyte cells, resulting in impaired memory B-cell and anti-polysaccharide antibody formation [18][19][20]. Also, the IgM antibody response to an influenza virus vaccine is impaired [20]. There are also lower T-cell subsets CD4+ and CD8+ in SCD patients, which affect their divergence into mature lymphocytes [21]. Therefore, there is decreased production in both the Th1 (IFN-gamma, IL-2, and TNF-beta) and Th2 (IL-4, IL-5, IL-6, IL-9, IL-10, and IL-13) responses of CD4+ T-helper cells in SCD [22].
2.5. Nutritional Deficiencies
Nutritional deficiencies also have a major impact on SCD children’s immune systems. SCD patients have macro-and micronutrient deficiencies due to pathways that include reduced calorie consumption, increased basal metabolic rate (BMR), increased RBC synthesis, increased protein turnover, dysregulated inflammatory response, and high myocardial energy requirements. Micronutrient deficiencies are associated with increased susceptibility to infection and a higher rate of SCD complications [23]. Low plasma zinc levels in malnourished children usually result in immune dysfunction. IL-2 is required for cell-mediated immunity maturation, and zinc deficiency causes lymphopenia, a decreased production of IL-2, which impairs the coordination of the innate and adaptive immune systems. Zinc deficiency is caused by insufficient food consumption, high protein turnover, and increased loss from the kidney due to insufficient reabsorption. Zinc supplementation improves somatic development in SCD children, and vitamins A, B, and magnesium have been shown to reduce inflammation, vaso-occlusive crisis, and hospitalizations [24][25][26][27][28].
2.6. Hereditary Influences
Despite having the same genetic defect, the severity of SCD varies widely among individuals because not all patients have identical pleiotropic genes. Some carriers have mutated genes, whichcan either improve or worsen the phenotype. This suggests that the phenotype of SCD is multigenic. Some other genes unrelated to the globin locus participate in relevant pathological events (rapid destruction of sickle cells, dense cell formation, endothelial adhesion) controlled by many, known as pleiotropic or secondary effector genes [29]. Polymorphisms in various immune-related genes have been proposed as a factor for increased vulnerability to infection in SCD patients. Previous findings show direct involvement of the HLA class II polymorphism in developing primary infections in SCD [30]. The increased risk of bacteremia may be controlled by the mannose-binding lectin receptor, Fc receptor, beta-S gene cluster haplotypes, IGF1R, and the TGF-beta/bone morphogenetic protein (BMP) pathway [31][32][33][34].
2.7. Mechanical Factors
Several mechanical factors may also play a role in a wide range of infections in SCD patients. Environmental factors can contribute to a variety of infections in SCD. The leading cause of respiratory infection in SCD is air pollution. Tobacco smoke exposure increases the risk of sickle cell crises in children with SCD due to chronic tissue hypoxia, vascular endothelial cell damage, and thrombus formation.
Furthermore, tobacco smoke may indirectly exacerbate asthma and acute chest syndrome in patients with SCD [35][36]. SCD patients are also predisposed to a variety of iatrogenic infections as a result of therapeutic interventions. Some of the complications treated by blood transfusion include vaso-occlusive crisis, splenic sequestration, acute chest syndrome, priapism, and strokes. Blood transfusions without pathogen screening can increase the risk of blood-borne infections, most notably hepatitis B and C, HIV, cytomegalovirus (CMV), and B19 parvovirus.
2.8. Molecular Mechanism of Clinical Manifestations in SCD
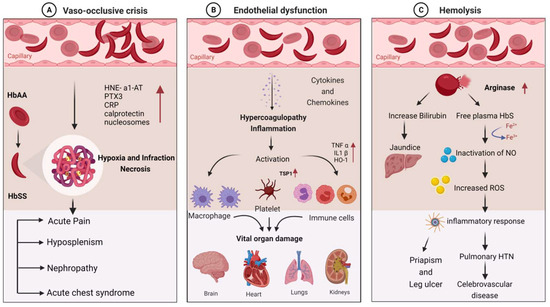
SCD patients are susceptible to infections due to a variety of immunological abnormalities and exposure to infectious agents. Pathophysiological mechanisms of the disease can explain the disease’s role in stimulating immune dysfunction. The clinical manifestations of infection in SCD are primarily vaso-occlusion, which causes endothelial dysfunction and hemolysis. The detailed molecular mechanism involved in the clinical manifestations of SCD is depicted in Figure 1.
Figure 1. Molecular mechanism involved in the clinical manifestations of SCD due to infections: (A) vaso-occlusive crisis; (B) endothelial dysfunction; (C) hemolysis.
Hematocrit, plasma viscosity, and erythrocyte deformability are some factors that affect blood rheology. Sickled RBCs become mechanically trapped in the microcirculation, promoting adhesive events among blood cells and resulting in chronic vaso-occlusion, causing frequent episodes of pain, hemolytic anemia, organ damage, and premature death [37][38]. Sickled RBCs also promote the exposure of adhesion molecules and binding motifs that are not generally found on RBCs’ outer membranes, such as phosphatidyl serine (PS), basal cell adhesion molecule-1 (B-CAM1), integrin-associated protein (IAP), and intercellular-adhesion-molecule-4 (ICAM-4). Sickled RBCs also have an integrin complex on their surface, which binds to fibronectin and vascular-cell adhesion molecule 1 (VCAM1), expressed on endothelial cells’ membrane and activated by inflammatory cytokines like tumor necrosis factor-alpha. C-reactive protein (CRP) is generated in response to interleukin-6 and pentraxin 3 (PTX3), generated mainly by neutrophils and endothelial cells as a result of an inflammatory response [39][40][41]. Thrombospondin, secreted by activated platelets, binds to endothelial cells and sickled RBC via CD36 and sulfated glycans found only on sickled RBC.
The interaction of sickled RBC with vascular endothelium may result in the formation of oxygen radicals by the endothelial cell and the transcription factor NF-kB by oxidants. When NF-kB is activated, the transcription of various genes of adhesion molecules, such as E- and P-selectin, VCAM-1, and ICAM-1, on the endothelial surface is increased [2]. It also increases the expression of major leukocyte chemo-attractants, such as interleukin-8 (IL-8) on endothelial cells [1][42]. Neutrophil-derived azurophilic protein elastase, with its inhibitor a1-antitrypsin (HNE-a1-AT), and neutrophil cytosolic protein calprotectin levels are increased with the activation of neutrophils. The inflammatory environment during VOC may also promote neutrophil, monocyte, and platelet activation, which increases their adhesion to each other and activated endothelial cells.
Hemolysis causes Hb to oxidize, resulting in the formation of heme and its oxidized form. The Fe3+ and Fe4+ states of hemoglobin formed in SCD are highly reactive in terms of encouraging oxidation. Hemolysis also results in red cell microparticles, which can deliver toxic heme to endothelial cells [43][44]. These hemolysis products are thought to be erythrocyte danger-associated molecular patterns (eDAMPs), which trigger innate immune responses and may play a role in sickle cell inflammation [45][46]. Hemin is also a potent TLR4 agonist, which contributes to a proinflammatory and procoagulant state in SCD. Because of the increased production of placental growth factor (PIGF) resulting from ischemia–reperfusion injury, monocytes are activated in response to PIGF, secreting more TNF-a, IL-1, and other chemokines. These increased oxidant production and leukocyte adhesion to the endothelium, followed by extravasation into the tissues and, finally, tissue damage [47].
2.9. Infectious Complications Associated with SCD
Researchers previously identified bacterial infections as the leading cause of morbidity and mortality in SCD patients, and children are the most vulnerable age group [48][49][50][51]. SCD increases susceptibility to various microbial, viral, parasitic, and Protozoan infections (Figure 2). Bacteremia/sepsis is caused by both Gram-positive (S. pneumonia and S. aureus) and Gram-negative (E. coli, Klebsiella, and Bacteroides) bacteria, which is the most common infection reported [52]. The infections become more severe during severe anemia and recurrent VOC. The prevalence, susceptibility, and severity of the effect on disease depend on the age group. Children are more susceptible to sepsis and pneumococcal infection [53].
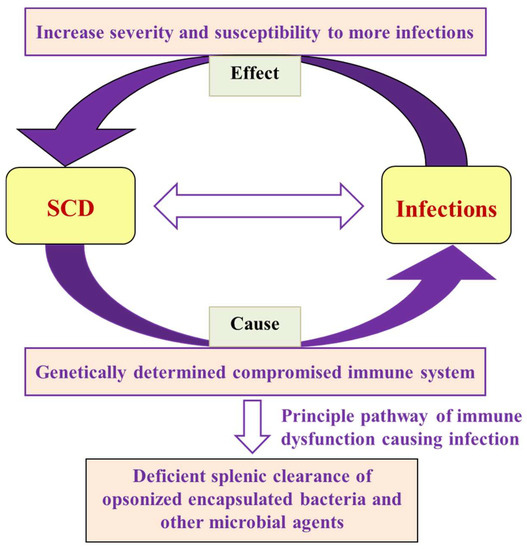
Figure 2. Flow chart showing the interrelationship of SCD and infections in terms of cause and effect, along with the failure of the primary mechanism that leads to microbial and parasitic infections.
2.10. Bacterial Infections
2.10.1. Bacteremia, Sepsis, and Pneumonia
Bacteremia and sepsis are common complications of SCD. Streptococcus pneumonia, Neisseria meningitides, Haemophilus influenza, and Escherichia coli cause sepsis [54]. Klebsiella pneumonia and Staphylococcus aureus are two other bacteria that can cause bacteremia and septicemia [55]. Salmonella species and Pantoea agglomerans enteric Gram-negative bacteria also negatively impact SCD patients [56][57]. An extensive data analysis of 550 SCD patients shows that the use of catheters during red cell exchange/transfusion increased the risk of bacteremia or thrombosis [58].
Children with SCD have immune dysfunctions that cause severe sepsis symptoms, which is less common in normal children [59]. Sickling infants are more susceptible to Streptococcus pneumonia infections due to several factors, including the interference of antibody production, opsonophagocytosis, and functional asplenia, as well as defective splenic clearance [60]. On the other hand, children and adults are more susceptible to S. pneumonia infections due to a lack of IgG and IgM antibody response and impairments in splenic complement and opsonophagocytic functions [54]. Penicillin prophylaxis and vaccination against bacteremia are preventive measures for infections that have reduced childhood mortality [61].
2.10.2. Acute Chest Syndrome (ACS)
Acute chest syndrome is one of the major clinical manifestations of SCD, leading to death. Primarily bacterial and viral infections cause ACS due to functional asplenia. Children under five years of age with SCD have been reported with high mortality rates because of Streptococcus pneumonia infection, responsible for ACS [62]. S. pneumonia is also a causative organism for severe anemia.
2.10.3. Meningitis
Recently Chenou et al. observed that SCD patients are 300 times more likely to develop bacterial meningitis than the normal population, and 10% of affected children die due to the infection generated by meningitis and pneumococcus [63]. Previous research found that meningitis in SCD patients caused by various bacteria, including H. influenzae, H. meningitis, and E. coli [64], and Streptococcus pneumonia, was responsible for 70% of bacterial meningitis cases in SCD children, while H. influenza was responsible for 80% [63][65]. The recurrence of meningitis occurs more frequently in SCD patients [66].
2.10.4. Osteomyelitis
Osteomyelitis is characterized by bone inflammation and is found in up to 61% of SCD patients, particularly in long bones such as the femur, humerus, and jaws. It is more common in SCD patients, and it is the second most affected tissue after the spleen [67]. The episodes of vaso-occlusive crisis ruptured the vasculature around the bones, allowing bacteria to infect the site, which is the primary cause of osteomyelitis [68]. Most SCD patients with osteomyelitis have Salmonella infections at multiple bone sites, which are most frequent in early childhood compared to non-SCD individuals [69][70].
2.10.5. Mycobacteria
It has been reported that iron overload is primarily known to cause organ dysfunction and also increases the risk for mycobacterial infection in SCD patients [71][72]. Shemisa et al. found that SCD Patients with iron overload were at an increased 17-fold higher risk to die from Mycobacterium tuberculosis in the sub-Saharan African population [73]. Thorell et al. reported that nontuberculous mycobacteria (NTM) infection, such as Mycobacterium fortuitum, causes frequent admissions for vaso-occlusive painful episodes in teenagers with SCD [74].
2.10.6. Urinary Tract Infection (UTI)
Urinary tract infection is a common cause of childhood morbidity and mortality in SCD. A high prevalence of UTIs with characteristics of pyelonephritis caused by Salmonella, Staphylococcus, Escherichia coli, and Enterobacter–Klebsiella was seen in SCD patients [75]. It was observed that scarring while healing the renal medulla and excretion of dilute urine were responsible for increased UTIs and pyelonephritis.
2.10.7. Gastrointestinal Infections
SCD patients have frequent hypoxia–reperfusion injuries caused by VOC in the intestinal vasculature, increasing gut permeability [76]. The accumulation of microbes and their products activates neutrophils, resulting in VOC [77]. The finding of ischemic colitis as an etiology of intestinal VOC has been observed in SCD patients [78].
2.11. Viral Infection
2.11.1. Respiratory Infections (RI)
Lung function abnormalities, or RI, are common in children and adults with SCD and may lead to a progressive decline in lung function with age [79][80]. The respiratory system is affected by several syncytial viruses, such as influenza, rhinoviruses, human metapneumo, and para-influenza, resulting in respiratory failure, which causes ACS [81]. The symptoms and severity are similar to seasonal influenza reported in SCD children and teenagers [82]. More hospitalization rates have been observed due to seasonal influenza infection being observed more in SCD children than normal [83].
2.11.2. Anemia Associated with Viral Infections
Severe anemia due to a transient aplastic crisis caused by B19 parvovirus belonging to the genus Erythrovirus (family Parvoviridae) was observed in 65–80% of SCD patients. It infects the erythroid progenitor cells and obstructs erythropoiesis [84]. B19 parvovirus has been associated with ACS, splenic and hepatic sequestration, bone marrow necrosis, pain crisis, and stroke [85].
2.11.3. Hepatitis B and C Infections
Both hepatitis B virus (HBV) and hepatitis C virus (HCV) infections have a common transmission mode and can cause chronic liver infection. The prevalence of hepatitis B infections is globally higher than hepatitis C, and the infection rate is higher in males than in females [86].
2.11.4. HIV Infections
There are many crosstalks between HIV and SCD; thus, the coexistence of HIV and SCD has a synergistic effect. HIV infection was found in 0–11.5% of SCD patients [87]. HIV infection also creates a favorable environment for pneumococcal infection, which can be fatal, resulting in severe pneumonia or meningitis in SCD adults [88]. A nested case-control study by Belisário et al. showed that HIV infection increased the severity of SCD-related symptoms such as ACS/pneumonia, sepsis/bacteremia, pyelonephritis, ischemic stroke, hemorrhagic stroke, abnormal transcranial doppler, and pulmonary hypertension [89].
2.11.5. Dengue Virus
Dengue virus is a mosquito-borne flavivirus that is most common in tropical and subtropical areas [90]. Dengue causes headaches, fever, abdominal pain, bleeding, myalgias, and loss of capillary integrity and increased mortality by up to 12.5% in SCD patients [91][92][93], due to their compromised immune systems, which make them more susceptible to hypovolemia and endothelial cell activation.
2.11.6. Coronavirus Disease (SARS-CoV-2)
Since 2019, SARS-CoV-2 coronavirus (COVID-19) infection has been a worldwide concern. Individuals with co-morbidities are more likely to become infected with the coronavirus. Individuals with HbSS hemoglobin have hypoxia or ACS. They are thought to be more susceptible to SARS-CoV-2 infection, which affects the lungs and respiratory tract. Case reports confirmed ACS and VOC complications in SCD patients with weakened immunity and infected with the novel coronavirus [94]. However, reports on the impact of COVID-19 in terms of severity and mortality are contradictory. Some African countries reported ACS and other co-morbidities in SCD patients with no casualties [95]. According to a survey of COVID-19 in hemoglobinopathy from the United Kingdom, the majority of patients hospitalized due to SARS-CoV-2 coronavirus infections had SCD (85.1%) and were primarily homozygous (Hb SS) patients (64.1%) [96].
2.12. Parasitic Infections
2.12.1. Malaria
Malaria is a highly vulnerable parasitic infection exhibiting fatal clinical manifestations, such as impaired splenic function in homozygous (HbSS) patients compared to HbAS or normal individuals. Individuals with HbAS show a protective effect and a lower frequency of malaria infection compared to normal individuals due to the HbAS making an unfavorable environment for malaria parasites. Moreover, the phagocytic action of infected RBC macrophages reduces infection rates [97][98][99][100][101]. Therefore, heterozygote individuals with the sickle cell gene are malaria-resistant compared to normal individuals. P. falciparum infection is also lower due to impaired erythrocyte membrane protein 1 (PfEMP 1) binding [102].
2.12.2. Other Parasitic Infections
In addition to Plasmodium, intestinal parasites such as Entamoeba histolytica, Entamoeba coli, and Giardia lamblia, and some helminths, such as Ascaris lumbricoides, Ancylostoma duodenale, Trichuris trichiura, and Strongyloides stercoralis, cause severe anemia in SCD patients.
2.13. Treatment of Infections in SCD
Bacterial infection and sepsis are common in SCD patients as their immune systems are compromised. These should be treated with broad-spectrum antibiotics, including third-generation cephalosporins like ceftriaxone, cefotaxime oxacillin, nafcillin, or cefazolin vancomycin, and clindamycin. Penicillin, beta-lactam inhibitors, and amphotericin B deoxycholate, along with third-generation cephalosporins should be used for meningitis.
Moreover, oral therapies are also given based on infecting parasite species. SCD patients are also very susceptible to different parasitic infections; common medicines, like albendazole, mebendazole, doxycycline, and ivermectin are given. Viral infections are very dangerous for SCD patients, as they are already suffering from other disease complications. Treatment options for viral diseases like HIV vary with antiretroviral drug resistance and adverse event profiles, and consultation with an HIV expert is recommended. The emergence of COVID-19 from SARS-CoV-2 also put SCD patients into life-threatening conditions. Hydroxyurea, L-glutamine, voxelotor, and crizanlizumab are the primary medications for preventing hemolysis and VOC.
3. Conclusions
Pneumonia, meningitis, osteomyelitis, and UTIs are prevalent among individuals with SCD in developing countries, constituting the leading factors behind its morbidity and mortality. It has also been observed that the primary infectious symptoms occur in children under the age of five, necessitating the need for immediate treatment. Priorities for the future management of infectious complications in SCD vary according to geography and socioeconomic status. Susceptibility to infection varies among individuals and across age groups. Some patients have chronic hemolytic anemia with vital organ failure due to infarctive damage, while others have no or few medical problems. A better understanding of the mechanisms underlying the increased susceptibility of these patients to infection may lead to interventions that address the underlying cause in the future. Meanwhile, the early detection and treatment of infections with hydroxyurea and blood transfusions can help to avoid severe complications and splenic dysfunction. Simultaneously, primary interventions, such as penicillin prophylaxis and vaccinations, result in significant improvements in SCD patients.
This entry is adapted from the peer-reviewed paper 10.3390/thalassrep13030019
References
- Kaul, D.K.; Finnegan, E.; Barabino, G.A. Sickle Red Cell–Endothelium Interactions. Microcirculation 2009, 16, 97–111.
- Sundd, P.; Gladwin, M.T.; Novelli, E.M. Pathophysiology of Sickle Cell Disease. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 263–292.
- Lakkakula, B.V.; Sahoo, R.; Verma, H.; Lakkakula, S. Pain Management Issues as Part of the Comprehensive Care of Patients with Sickle Cell Disease. Pain Manag. Nurs. 2018, 19, 558–572.
- Patra, P.K.; Lakkakula, B.V.K.S.; Verma, H.K.; Choubey, M.; Patra, S.; Khodiar, P.K. Assessment of renal function in Indian patients with sickle cell disease. Saudi J. Kidney Dis. Transplant. 2017, 28, 524–531.
- Bhaskar, L.V.K.S.; Shukla, P.; Verma, H.; Patel, S.; Patra, P. Ocular manifestations of sickle cell disease and genetic susceptibility for refractive errors. Taiwan J. Ophthalmol. 2017, 7, 89–93.
- Verma, H.K.; Lakkakula, S.; Lakkakula, B.V. Retrospection of the effect of hydroxyurea treatment in patients with sickle cell disease. Acta Haematol. Pol. 2018, 49, 1–8.
- Jha, A.N.; Mishra, H.; Verma, H.K.; Pandey, I. Compound Heterozygosity of beta-Thalassemia and the Sickle Cell Hemoglobin in Various Populations of Chhattisgarh Statel, India. Hemoglobin 2018, 2, 84–90.
- Bhanushali, A.A.; Patra, P.; Nair, D.; Verma, H.; Das, B. Genetic variant in the BCL11A (rs1427407), but not HBS1-MYB (rs6934903) loci associate with fetal hemoglobin levels in Indian sickle cell disease patients. Blood Cells Mol. Dis. 2015, 54, 4–8.
- Al-Salem, A.H. Splenic Complications of Sickle Cell Anemia and the Role of Splenectomy. ISRN Hematol. 2011, 2011, 864257.
- Monaco, C.P.; Fonseca, P.B.B.; Braga, J.A.P. Infectious complications after surgical splenectomy in children with sickle cell disease. Rev. Paul. Pediatr. (Engl. Ed.) 2015, 33, 150–153.
- Hsu, L.; E Nnodu, O.; Brown, B.J.; Tluway, F.; King, S.; Dogara, L.G.; Patil, C.; Shevkoplyas, S.S.; Lettre, G.; Cooper, R.S.; et al. White Paper: Pathways to Progress in Newborn Screening for Sickle Cell Disease in Sub-Saharan Africa. J. Trop. Dis. 2018, 6, 260.
- Bohnsack, J.F.; Brown, E.J. The role of the spleen in resistance to infection. Annu. Rev. Med. 1986, 37, 49–59.
- Ansari, J.; Gavins, F.N.E. Ischemia-Reperfusion Injury in Sickle Cell Disease: From Basics to Therapeutics. Am. J. Pathol. 2019, 189, 706–718.
- West, T.B.; West, D.W.; Ohene-Frempong, K. The presentation, frequency, and outcome of bacteremia among children with sickle cell disease and fever. Pediatr. Emerg. Care 1994, 10, 141–143.
- Halasa, N.B.; Shankar, S.M.; Talbot, T.R.; Arbogast, P.G.; Mitchel, E.F.; Wang, W.C.; Schaffner, W.; Craig, A.S.; Griffin, M.R. Incidence of Invasive Pneumococcal Disease among Individuals with Sickle Cell Disease before and after the Introduction of the Pneumococcal Conjugate Vaccine. Clin. Infect. Dis. 2007, 44, 1428–1433.
- Dunkelberger, J.R.; Song, W.-C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50.
- Spirer, Z.; Weisman, Y.; Zakuth, V.; Fridkin, M.; Bogair, N. Decreased serum tuftsin concentrations in sickle cell disease. Arch. Dis. Child. 1980, 55, 566–567.
- Cameron, P.U.; Jones, P.; Gorniak, M.; Dunster, K.; Paul, E.; Lewin, S.; Woolley, I.; Spelman, D. Splenectomy Associated Changes in IgM Memory B Cells in an Adult Spleen Registry Cohort. PLoS ONE 2011, 6, e23164.
- Weller, S. Human blood IgM “memory” B cells are circulating splenic marginal zone B cells harboring a prediversified immunoglobulin repertoire. Blood 2004, 104, 3647–3654.
- Ballester, O.F.; Abdallah, J.M.; Prasad, A.S. Impaired IgM antibody responses to an influenza virus vaccine in adults with sickle cell anemia. Am. J. Hematol. 1985, 20, 409–412.
- Koffi, K.G.; Sawadogo, D.; Meite, M.; Nanho, D.C.; Tanoh, E.S.; Attia, A.K.; Sanogo, I.; Sangare, A. Reduced levels of T-cell subsets CD4+ and CD8+ in homozygous sickle cell anaemia patients with splenic defects. Hematol. J. 2003, 4, 363–365.
- Romagnani, S. T-cell subsets (Th1 versus Th2). Annals of allergy, asthma & immunology: Official publication of the American College of Allergy. Asthma Immunol. 2000, 85, 9–18.
- Hibbert, J.M.; Creary, M.S.; E Gee, B.; Buchanan, I.D.; Quarshie, A.; Hsu, L.L. Erythropoiesis and Myocardial Energy Requirements Contribute to the Hypermetabolism of Childhood Sickle Cell Anemia. J. Pediatr. Gastroenterol. Nutr. 2006, 43, 680–687.
- Prasad, A.S.; Schoomaker, E.B.; Ortega, J.; Brewer, G.J.; Oberleas, D.; Oelshlegel, F.J. Zinc Deficiency in Sickle Cell Disease. Clin. Chem. 1975, 21, 582–587.
- Prasad, A.S. Zinc deficiency and effects of zinc supplementation on sickle cell anemia subjects. Prog. Clin. Biol. Res. 1981, 55, 99–122.
- Fraker, P.J.; King, L.E.; Laakko, T.; Vollmer, T.L. The Dynamic Link between the Integrity of the Immune System and Zinc Status. J. Nutr. 2000, 130, 1399S–1406S.
- De Franceschi, L.; Bachir, D.; Galacteros, F.; Tchernia, G.; Cynober, Y.; Neuberg, D.; Beuzard, Y.; Brugnara, C. Oral magnesium pidolate: Effects of long-term administration in patients with sickle cell disease. Br. J. Haematol. 2000, 108, 284–289.
- Schall, J.I.; Zemel, B.S.; Kawchak, D.A.; Ohene-Frempong, K.; Stallings, V.A. Vitamin A status, hospitalizations, and other outcomes in young children with sickle cell disease. J. Pediatr. 2004, 145, 99–106.
- Stuart, M.J.; Nagel, R.L. Sickle-cell disease. Lancet 2004, 9442, 1343–1360.
- Tamouza, R.; Neonato, M.-G.; Busson, M.; Marzais, F.; Girot, R.; Labie, D.; Elion, J.; Charron, D. Infectious complications in sickle cell disease are influenced by HLA class II alleles. Hum. Immunol. 2002, 63, 194–199.
- Neonato, M.-G.; Lu, C.Y.; Guilloud-Bataille, M.; Lapouméroulie, C.; Nabeel-Jassim, H.; Dabit, D.; Girot, R.; Krishnamoorthy, R.; Feingold, J.; Besmond, C.; et al. Genetic polymorphism of the mannose-binding protein gene in children with sickle cell disease: Identification of three new variant alleles and relationship to infections. Eur. J. Hum. Genet. 1999, 7, 679–686.
- Norris, C.F.; Surrey, S.; Bunin, G.R.; Schwartz, E.; Buchanan, G.R.; McKenzie, S.E. Relationship between Fc receptor IIA polymorphism and infection in children with sickle cell disease. J. Pediatr. 1996, 128, 813–819.
- Powars, D.R.; Chan, L.; Schroeder, W.A. Beta S-gene-cluster haplotypes in sickle cell anemia: Clinical implications. Am. J. Pediatr. Hematol. Oncol. 1990, 12, 367–374.
- Adewoye, A.H.; Nolan, V.G.; Ma, Q.; Baldwin, C.; Wyszynski, D.F.; Farrell, J.J.; Farrer, L.A.; Steinberg, M.H. Association of polymorphisms of IGF1R and genes in the transforming growth factor-beta/bone morphogenetic protein pathway with bacteremia in sickle cell anemia. Clin. Infect. Dis.Off. Publ. Infect. Dis. Soc. Am. 2006, 43, 593–598.
- West, D.C.; Romano, P.S.; Azari, R.; Rudominer, A.; Holman, M.; Sandhu, S. Impact of Environmental Tobacco Smoke on Children With Sickle Cell Disease. Arch. Pediatr. Adolesc. Med. 2003, 157, 1197–1201.
- Cohen, R.T.; DeBaun, M.R.; Blinder, M.A.; Strunk, R.C.; Field, J.J. Smoking is associated with an increased risk of acute chest syndrome and pain among adults with sickle cell disease. Blood 2010, 115, 3852–3854.
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-cell disease. Lancet 2010, 376, 2018–2031.
- Cisneros, G.S.; Thein, S.L. Recent Advances in the Treatment of Sickle Cell Disease. Front. Physiol. 2020, 11, 435.
- Krishnan, S.; Setty, Y.; Betal, S.G.; Vijender, V.; Rao, K.; Dampier, C.; Stuart, M. Increased levels of the inflammatory biomarker C-reactive protein at baseline are associated with childhood sickle cell vasocclusive crises. Br. J. Haematol. 2010, 148, 797–804.
- Bargoma, E.M.; Mitsuyoshi, J.K.; Larkin, S.K.; Styles, L.A.; Kuypers, F.A.; Test, S.T. Serum C-reactive protein parallels secretory phospholipase A2 in sickle cell disease patients with vasoocclusive crisis or acute chest syndrome. Blood 2005, 105, 3384–3385.
- Jaillon, S.; Peri, G.; Delneste, Y.; Frémaux, I.; Doni, A.; Moalli, F.; Garlanda, C.; Romani, L.; Gascan, H.; Bellocchio, S.; et al. The humoral pattern recognition receptor PTX3 is stored in neutrophil granules and localizes in extracellular traps. J. Exp. Med. 2007, 204, 793–804.
- Kato, G.J.; Steinberg, M.H.; Gladwin, M.T. Intravascular hemolysis and the pathophysiology of sickle cell disease. J. Clin. Investig. 2017, 127, 750–760.
- Camus, S.M.; De Moraes, J.A.; Bonnin, P.; Abbyad, P.; Le Jeune, S.; Lionnet, F.; Loufrani, L.; Grimaud, L.; Lambry, J.-C.; Charue, D.; et al. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood 2015, 125, 3805–3814.
- Reeder, B.J.; Andersen, C.B.F.; Stødkilde, K.; Sæderup, K.L.; Kuhlee, A.; Raunser, S.; Graversen, J.H.; Moestrup, S.K.; Strader, M.B.; Alayash, A.I.; et al. The Redox Activity of Hemoglobins: From Physiologic Functions to Pathologic Mechanisms. Antioxid. Redox Signal. 2010, 13, 1087–1123.
- Gladwin, M.T.; Ofori-Acquah, S.F. Erythroid DAMPs drive inflammation in SCD. Blood 2014, 123, 3689–3690.
- Mendonça, R.; Silveira, A.A.A.; Conran, N. Red cell DAMPs and inflammation. Inflamm. Res. 2016, 65, 665–678.
- Mpollo, M.-S.E.M.; Brandt, E.B.; Shanmukhappa, S.K.; Arumugam, P.I.; Tiwari, S.; Loberg, A.; Pillis, D.; Rizvi, T.; Lindsey, M.; Jonck, B.; et al. Placenta growth factor augments airway hyperresponsiveness via leukotrienes and IL-13. J. Clin. Investig. 2015, 126, 571–584.
- Onwubalili, J.K. Sickle cell disease and infection. J. Infect. 1983, 7, 2–20.
- Adeyokunnu, A.; Hendrickse, G. Salmonella osteomyelitis in childhood. Arch. Dis. Child. 1980, 55, 175–184.
- El-Hazmi, M. Infections in Sickle Cell Disease. Ann. Saudi Med. 1986, 6, 33–40.
- Powars, D.; Overturf, G.; Turner, E. Is There an Increased Risk of Haemophilus influenzae Septicemia in Children with Sickle Cell Anemia? Pediatrics 1983, 71, 927–931.
- Ochocinski, D.; Dalal, M.; Black, L.V.; Carr, S.; Lew, J.; Sullivan, K.; Kissoon, N. Life-Threatening Infectious Complications in Sickle Cell Disease: A Concise Narrative Review. Front. Pediatr. 2020, 8, 38.
- Ahmed, S.G. The Role of Infection in the Pathogenesis of Vaso-Occlusive Crisis in Patients with Sickle Cell Disease. Mediterr. J. Hematol. Infect. Dis. 2011, 3, e2011028.
- Gonzales, J.; Chakraborty, T.; Romero, M.; Abu Mraheil, M.; Kutlar, A.; Pace, B.; Lucas, R. Streptococcus pneumoniae and Its Virulence Factors H2O2 and Pneumolysin Are Potent Mediators of the Acute Chest Syndrome in Sickle Cell Disease. Toxins 2021, 13, 157.
- Brown, B.; Dada-adegbola, H.; Trippe, C.; Olopade, O. Prevalence and Etiology of Bacteremia in Febrile Children with Sickle Cell Disease at a Nigeria Tertiary Hospital. Mediterr. J. Hematol. Infect. Dis. 2017, 9, 3–10.
- Wierenga, K.J.J.; Hambleton, I.R.; Wilson, R.M.; Alexander, H.; E Serjeant, B.; Serjeant, G.R. Significance of fever in Jamaican patients with homozygous sickle cell disease. Arch. Dis. Child. 2001, 84, 156–159.
- Al Achkar, M.; Rogers, J.S.; Muszynski, M.J. Pantoea species sepsis associated with sickle cell crisis in a pregnant woman with a history of pica. Am. J. Case Rep. 2012, 13, 26–28.
- Alkindi, S.; Matwani, S.; Al-Maawali, A.; Al-Maskari, B.; Pathare, A. Complications of PORT-A-CATH® in patients with sickle cell disease. J. Infect. Public Heal. 2012, 5, 57–62.
- Quinn, C.T. Sickle cell disease in childhood: From newborn screening through transition to adult medical care. Pediatr. Clin. North. Am. 2013, 60, 1363–1381.
- Battersby, A.J.; Knox-Macaulay, H.H.; Carrol, E.D. Susceptibility to invasive bacterial infections in children with sickle cell disease. Pediatr. Blood Cancer 2010, 55, 401–406.
- Rincón-López, E.M.; Gómez, M.L.N.; Matos, T.H.; Saavedra-Lozano, J.; de la Red, Y.A.; Rupérez, B.H.; de Julián, E.C. RETRO-DREP Study Group Low-risk factors for severe bacterial infection and acute chest syndrome in children with sickle cell disease. Pediatr. Blood Cancer 2019, 66, e27667.
- Wahl, B.; Brien, K.L.O.; Greenbaum, A.; Majumder, A.; Liu, L.; Chu, Y.; Lukšić, I.; Nair, H.; McAllister, D.A.; Campbell, H.; et al. Articles Burden of Streptococcus pneumoniae and Haemophilus influenzae type b disease in children in the era of conjugate vaccines: Global, regional, and national estimates for. Lancet Glob. Health 2018, 6, e744–e757.
- Chenou, F.; Azevedo, J.; Leal, H.F.; Gonçalves, M.d.S.; Reis, J.N. Bacterial meningitis in patients with sickle cell anemia in Salvador, Bahia, Brazil: A report on ten cases. Hematol. Transfus. Cell Ther. 2020, 42, 139–144.
- Kiriazopulos, D.; Pedroni, P.; Occhi, G.; Sassi, G.; Corna, A.; Cieri, F.; Cipolletta, E.; Orobello, M.; Colombo, M. Pneumococcal Meningitis in a Child With Sickle Cell Anemia: A Case Report. Int. J. Clin. Pediatr. 2015, 4, 168–170.
- Nottidge, V.A. Pneumococcal Meningitis in Sickle Cell Disease in Childhood. Arch. Pediatr. Adolesc. Med. 1983, 137, 29–31.
- Coyle, P. Overview of Acute and Chronic Meningitis. Neurol. Clin. 1999, 17, 691–710.
- Junior, G.B.D.S.; Daher, E.D.F.; Da Rocha, F.A.C. Osteoarticular involvement in sickle cell disease. Rev. Bras. Hematol. E Hemoter. 2012, 34, 156–164.
- Mary, P. Sickle cell disease as a cause of osteoarthritis. Arch. Pediatr. 2008, 15, 639–641.
- Anand, A.J.; Glatt, A.E. Salmonella osteomyelitis and arthritis in sickle cell disease. Semin. Arthritis Rheum. 1994, 24, 211–221.
- AlFawaz, T.; Alzumar, O.; AlShahrani, D.; Alshehri, M. Severity of Salmonella infection among sickle cell diseases pediatric patients: Description of the infection pattern. Int. J. Pediatr. Adolesc. Med. 2019, 6, 115–117.
- Coates, T.D.; Wood, J.C. How we manage iron overload in sickle cell patients. Br. J. Haematol. 2017, 177, 703–716.
- Raghupathy, R.; Manwani, D.; Little, J.A. Iron Overload in Sickle Cell Disease. Adv. Hematol. 2010, 2010, 272940.
- Shemisa, K.; Jafferjee, N.; Thomas, D.; Jacobs, G.; Meyerson, H.J. Mycobacterium avium Complex Infection in a Patient with Sickle Cell Disease and Severe Iron Overload. Case Rep. Infect. Dis. 2014, 2014, 405323.
- Thorell, E.A.; Sharma, M.; Jackson, M.A.; Selvarangan, R.; Woods, G.M. Disseminated Nontuberculous Mycobacterial Infections in Sickle Cell Anemia Patients. J. Pediatr. Hematol. 2006, 28, 678–681.
- Yanda, A.N.A.; Nansseu, J.R.N.; Awa, H.D.M.; Tatah, S.A.; Seungue, J.; Eposse, C.; Koki, P.O.N. Burden and spectrum of bacterial infections among sickle cell disease children living in Cameroon. BMC Infect. Dis. 2017, 17, 211.
- Dutta, D.; Methe, B.; Amar, S.; Morris, A.; Lim, S.H. Intestinal injury and gut permeability in sickle cell disease. J. Transl. Med. 2019, 17, 183.
- Lim, S.H.; Fast, L.; Morris, A. Sickle cell vaso-occlusive crisis: It’s a gut feeling. J. Transl. Med. 2016, 14, 334.
- Green, B.T.; Branch, M.S. Ischemic colitis in a young adult during sickle cell crisis: Case report and review. Gastrointest. Endosc. 2003, 57, 605–607.
- MacLean, J.E.; Atenafu, E.; Kirby-Allen, M.; MacLusky, I.B.; Stephens, D.; Grasemann, H.; Subbarao, P. Longitudinal decline in lung volume in a population of children with sickle cell disease. Am. J. Respir. Crit. Care Med. 2009, 178, 1055–1059.
- Lunt, A.; McGhee, E.; Sylvester, K.; Rafferty, G.; Dick, M.; Rees, D.; Height, S.; Thein, S.L.; Greenough, A. Longitudinal assessment of lung function in children with sickle cell disease. Pediatr. Pulmonol. 2015, 51, 717–723.
- Claudio, A.M.; Foltanski, L.; Delay, T.; Britell, A.; Duckett, A.; Weeda, E.R.; Bohm, N. Antibiotic Use and Respiratory Pathogens in Adults With Sickle Cell Disease and Acute Chest Syndrome. Ann. Pharmacother. 2019, 53, 991–996.
- Alkindi, S.; Al-Yahyai, T.; Raniga, S.; Boulassel, M.R.; Pathare, A. Respiratory Viral Infections in Sickle Cell Anemia: Special Emphasis on H1N1 Co-infection. Oman Med. J. 2020, 35, e197.
- Bundy, D.G.; Strouse, J.J.; Casella, J.F.; Miller, M.R. Burden of influenza-related hospitalizations among children with sickle cell disease. Pediatrics 2010, 125, 234–243.
- Slavov, S.N.; Kashima, S.; Pinto, A.C.S.; Covas, D.T. Human parvovirus B19: General considerations and impact on patients with sickle-cell disease and thalassemia and on blood transfusions. FEMS Immunol. Med. Microbiol. 2011, 62, 247–262.
- Novelli, E.M.; Gladwin, M.T. Crises in Sickle Cell Disease. Chest 2016, 149, 1082–1093.
- Bakarey, A.S.; Akinboade, I.O.; Aken’ova, Y.A. Transmission transmissible hepatitis B virus markers of infection among sickle cell disease patients receiving care at a tertiary health facility in Ibadan, southwest Nigeria. J. Immunoass. Immunochem. 2018, 39, 416–427.
- Owusu, E.D.A.; Visser, B.J.; Nagel, I.M.; Mens, P.F.; Grobusch, M.P. The Interaction Between Sickle Cell Disease and HIV Infection: A Systematic Review. Clin. Infect. Dis. 2014, 60, 612–626.
- Robinson, T.M.; Lanzkron, S.M. Standard Definitions of Pneumococcal Immunity May Not Accurately Predict Protection in Adults with Sickle Cell Disease. Blood 2019, 134, 1014.
- Belisário, A.R.; Blatyta, P.F.; Vivanco, D.; Oliveira, C.D.L.; Carneiro-Proietti, A.B.; Sabino, E.C.; de Almeida-Neto, C.; Loureiro, P.; Mateos, S.d.O.G.; Flor-Park, M.V.; et al. Association of HIV infection with clinical and laboratory characteristics of sickle cell disease. BMC Infect. Dis. 2020, 20, 638.
- Halstead, S.; Wilder-Smith, A. Severe dengue in travellers: Pathogenesis, risk and clinical management. J. Travel Med. 2019, 26, taz062.
- Martina, B.E.E.; Koraka, P.; Osterhaus, A.D.M.E. Dengue Virus Pathogenesis: An Integrated View. Clin. Microbiol. Rev. 2009, 22, 564–581.
- Spiropoulou, C.F.; Srikiatkhachorn, A. The role of endothelial activation in dengue hemorrhagic fever and hantavirus pulmonary syndrome. Virulence 2013, 4, 525–536.
- Rankine-mullings, A.; Reid, M.E.; Moo, M.; Richards-dawson, M.; Knight, J.M. A Retrospective Analysis of the Signi fi cance of Haemoglobin SS and SC in Disease Outcome in Patients With Sickle Cell Disease and Dengue Fever. EBioMedicine 2015, 2, 937–941.
- Elia, G.M.; Angel, A.; Regacini, R.; Nais, R.P.; dos Santos, A.R.A.; Vieira, P.P.M.G.; Braga, J.A.P. Acute chest syndrome and COVID-19 in sickle cell disease pediatric patients. Hematol. Transfus. Cell Ther. 2020, 43, 104–108.
- Garg, S.; Kim, L.; Whitaker, M.; O’Halloran, A.; Cummings, C.; Holstein, R.; Prill, M.; Chai, S.J.; Kirley, P.D.; Alden, N.B.; et al. Hospitalization Rates and Characteristics of Patients Hospitalized with Laboratory-Confirmed Coronavirus Disease 2019—COVID-NET, 14 States, March 1–30, 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 458–464.
- Telfer, P.; De La Fuente, J.; Sohal, M.; Brown, R.; Eleftheriou, P.; Roy, N.; Piel, F.B.; Chakravorty, S.; Gardner, K.; Velangi, M.; et al. Real-time national survey of COVID-19 in hemoglobinopathy and rare inherited anemia patients. Haematologica 2020, 105, 2651–2654.
- Luzzatto, L. Sickle Cell Anaemia and Malaria. Mediterr. J. Hematol. Infect. Dis. 2012, 4, e2012065.
- Mwaiswelo, R.O.; Mawala, W.; Iversen, P.O.; de Montalembert, M.; Luzzatto, L.; Makani, J. Sickle cell disease and malaria: Decreased exposure and asplenia can modulate the risk from Plasmodium falciparum. Malar. J. 2020, 19, 165.
- Achkar, M.A.; Rogers, J.S.; Muszynski, M.J. Resistance to Plasmodium falciparum in sickle cell trait erythrocytes is driven by oxygen-dependent growth inhibition. Proc. Natl. Acad. Sci. USA 2018, 115, 7350–7355.
- Dada-Adegbola, H.O.; Brown, B.J.; Labaeka, A.A. Prevalence of malaria and performance of a rapid diagnostic test for malaria in febrile children with sickle cell disease. Pediatr. Hematol. Oncol. J. 2018, 3, 42–45.
- Goheen, M.; Wegmüller, R.; Bah, A.; Darboe, B.; Danso, E.; Affara, M.; Gardner, D.; Patel, J.; Prentice, A.; Cerami, C. Anemia Offers Stronger Protection Than Sickle Cell Trait Against the Erythrocytic Stage of Falciparum Malaria and This Protection Is Reversed by Iron Supplementation. EBioMedicine 2016, 14, 123–130.
- Cholera, R.; Brittain, N.J.; Gillrie, M.R.; Lopera-Mesa, T.M.; Diakité, S.A.S.; Arie, T.; Krause, M.A.; Guindo, A.; Tubman, A.; Fujioka, H.; et al. Impaired cytoadherence of Plasmodium falciparum -infected erythrocytes containing sickle hemoglobin. Proc. Natl. Acad. Sci. USA 2008, 105, 991–996.
This entry is offline, you can click here to edit this entry!