Gels are understood as soft viscoelastic multicomponent solids that are in the incomplete phase separation state, which, under the action of external mechanical forces, do not transit into a fluid state but rupture like any solid material. Gels can “melt” (again, like any solids) due to a change in temperature or variation in the environment. In contrast to this type of rheology, yielding liquids (sometimes not rigorously referred to as “gels”, especially in relation to colloids) can exist in a solid-like (gel-like) state and become fluid above some defined stress and time conditions (yield stress). At low stresses, their behavior is quite similar to that of permanent solid gels, including the frequency-independent storage modulus. The gel-to-sol transition considered in colloid chemistry is treated as a case of yielding. However, in many cases, the yield stress cannot be assumed to be a physical parameter since the solid-to-liquid transition happens in time and is associated with thixotropic effects.
1. Introduction
The term “gels” is one of the most widely used when scholars study various soft matters. The concept of “soft matter” was first proposed by deGennes when considering complex fluids and soft condensed matter [
1]. These matters have two main characteristics: a complex structure and flexibility. It is intuitively clear that under this definition, a certain group of multicomponent and, most likely, multiphase materials characterized by a low elastic modulus and, possibly, but not necessarily, fluidity is considered. The rheological properties of such systems should belong to the class of viscoplastic (or yielding) media since the dispersed components of the mixture should form a certain “structure” characterized by some strength. The term “structure” does not have a strict definition either since we are talking not only about crystalline bodies but also amorphous ones characterized by some specific interaction, causing the ordering of their parts and/or orientation in space. Soft matter is widely used in production, such as tensides, polymeric fluid compounds, liquid crystals, membranes, gels, concentrated emulsions, foams and other colloids, and proteins [
2].
The following terminological definitions will be adhered to:
- Gels are solid bodies incapable of irreversible deformations;
- Yielding liquids are materials that can be in a solid-like state but under some threshold conditions can transfer to a liquid state;
- A gel-like state is the solid-like state of a yielding liquid.
2. Gels and Gel-like States
2.1. Formation of a Gel
A gel is a multicomponent system formed by a structure-forming component and an absorbed liquid, usually a low-viscosity solvent. In particular, hydrogels can be defined as highly hydrophilic three-dimensional (3D) networks composed of cross-linked natural or synthetic polymer chains capable of holding huge amounts of water (>90%) [
9].
The general thermodynamic understanding of such compositions is “systems with incomplete phase separation” [
10]. This incompleteness is due to steric and/or kinetic reasons (
Figure 1):
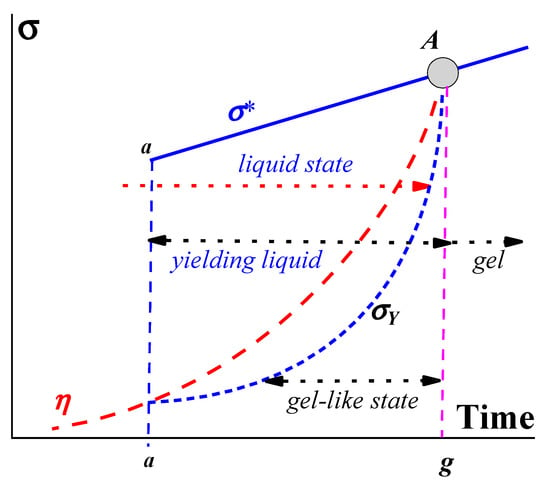
Figure 1. A rheological model of gelation.
The viscosity, η, of the initial composition of a liquid begins to grow due to its own intermolecular reactions, and at time a (in Figure 1), molecular interactions lead to the formation of a structure characterized by the yield stress, σY. This corresponds to the transition from a liquid (fluid) state to a yielding state, which exists during the time interval a–g. This interval consists of two parts: a gel-like state (at stresses below σY) and a liquid state (at stresses above σY), as shown by the arrows in Figure 1. The substance in a liquid state can demonstrate different rheological behaviors, including viscous flow and/or viscoelastic deformations, depending on the applied stress and time of its action. The upper limit in Figure 1 is the stress σ* (either shear or normal) at which the liquid is broken up. It should not be surprising that a liquid is characterized by a certain strength since complex liquids are always more or less elastic. Then, the destruction of such a liquid (as well as a gel), like any solid, is associated with the release of stored elastic energy. Thus, there are two characteristic parameters of the matter–macro-strength, σ*, and yield stress, σY. In the process of gelation, both of them increase over time and, at some point, become equal at point A. This means that the object in question becomes a real non-flowing gel.
At this point, it is necessary to define the difference between gels and yielding liquids. Indeed, the term “gel” is often used by scholars in various senses. According to the definition, a gel is a non-fluid soft elastic substance with a permanent structure, and the application of external forces results in viscoelastic reversible deformations, which, when a certain critical stress state (at
σ*) is reached, are destroyed. At the same time, there are many multicomponent compositions, e.g., polymer solutions with a transient structure that can exist as viscoelastic solids and become yielding fluids when an applied load exceeds some threshold. Such media (viscoplastic substances according to the standard but possibly not quite exact rheological nomenclature) are also frequently called “gels”. This extending using the concept of gels is popular, especially in colloid science, where yielding colloidal substances are usually treated as gels [
11,
12].
Meanwhile, a boundary between solutions and gels may not be rigid but somewhat blurry. It depends on the nature (energy and lifetime) of the intermolecular interactions. The characteristic times of the intermolecular contacts are determined by their energy and the energy of the Brownian motion. These times can be less than the time of observation (measurements), and in this case, it is a real solution. However, if components of the dispersed phase can create long-term associates and their lifetimes lie in the range acceptable for measurements, we arrive at the transition to the temporal gel state.
True gel (or solid gel), therefore, means exclusively a non-fluid solid substance. Such a gel may transit into a fluid state only when environmental changes (temperature, pH, etc.) occur. This is true, for example, for so-called “thermoreversible gels” [
13].
A substance that can be in a solid-like state and, under external forces, turn into a fluid state is called a “yielding liquid” (or “yielding stress” material). These substances (by analogy with Newtonian liquids) can also be called Binghamian liquids or media. The boundary between these two states of soft matter is determined by the yield stress. At stresses lower than the yield stress, such matter is considered as being in a gel-like or solid-like state, and at stresses higher than this threshold, the matter is considered to be in a liquid state, or sol. The transition between these states is traditionally (in colloid science) called a “gel-sol” (or vice versa) transition, although, in fact, it should be referred to as a gel-like (not gel) to sol transition.
Gels (solid-like gels), as a rule, are chemically cross-linked substances, the structural network of which is formed by covalent or ionic bonds. Due to the nature of these bonds, they are not able to perform the gel–sol transition under the applied external load. Yielding materials (liquids) are formed by the transient physical cross-linked network existing due to dispersed non-covalent interactions. The three-dimensional network of such a system can be reversibly (or partly irreversibly) destroyed by a high enough external load, and this is considered a gel–sol transition. This difference between the two types of rheological behavior is associated with categorizing gels into chemical gels and physical gels, where the former have permanent covalent bonds while the latter, like colloidal gels, have temporary and weak bonds [
14].
The term “plastic” in the mechanics of solids is understood as an irreversible deformation when this deformation depends on stress only, but at a certain stress, strain does not depend on time. That is why plastic deformations are something different to flow.
The creation of a solid-like structure, within which a large amount of liquid is retained, is widespread in various technologies. Important examples of such technology include the adsorption of crude oil mixed with water and the excess water production in petroleum reservoirs [
15,
16,
17,
18], hygroscopic polymer gels [
19], the development of stimuli-responsive material [
19,
20], 3D printing [
21], medicine and food soft matters [
22,
23], and fiber spinning from polymer solutions. The latter case is based on the effect of the phase separation of a polymer solution jet on contact with a coagulant. The state of the system is described by Equation (1) for the Flory–Huggins parameter:
where R is the gas constant, T is temperature, x is the ratio of molar volumes of polymer and solvent, and μ1 is the chemical potential (or the parameter of polymer–solvent interaction), which does not depend on φ2 and is proportional to 1/T.
One of the interesting and industrially used polymer gels is formed by the gel method of ultra-high molecular weight polyethylene processing into fibers, where the jet of the solution in non-polar solvent transforms to gel-like fiber under a decrease of temperature [
24].
Sorbents of various types are typical examples of industrial gels; among them, hyper-cross-linked co-polymers of styrene and divinylbenzene are special gel materials. These microporous gels have a free surface of more than 1000 m
2/g and can absorb a huge amount of liquid [
25,
26]. The development of new gels/sorbents continues due to the requirements of environmental safety and the oil industry, in particular. New super-absorbing cross-linked hydrogels are characterized by a swelling ratio exceeding 10 (at room temperature) [
27]. Mesoporous hydrogels also belong to the group of gels with a developed surface that can be important for biomedical applications [
28].
2.2. Rheology of Gels
Gels are materials formed at poin A in Figure 1 and possibly continuing to develop after point A. The X-axis may not necessarily represent time but, for example, temperature or a change in the composition of the matrix due to the addition of certain chemicals. According to the assumed definition, a gel is a solid matter without the ability to flow. The nature of the mechanical rupture of gels resembles the known mechanisms of breaking up of solids.
Structural bonds in a gel can be of different nature [
36]. The limiting case is strong covalent chemical bonds between macromolecules (chemical network), and the upper limit of such gels is plasticized rubbers and plastics (such as, for example, plasticized PVC and PVC plastisols). Their rheological behavior is quite well known. In the ideal case, it is characterized by a wide rubber-like plateau on the frequency dependence of the storage modulus
G′ (
ω) and relatively low values of the loss modulus
G″ (
ω).
The characteristics of gels on breaking up, as for other solid-like materials, have many similar features. First of all, two main types of break can be distinguished: brittle rupture and elastic yielding. The latter is intrinsic for polymeric substances [
37,
38], although brittle rupture has also been observed for these substances [
39].
Macro-fracture of gels (as well as other soft matters) can be observed in two modes–either as the appearance of discontinuities inside the sample due to the breaking of cohesive contact or the transition to wall slip instead of shear in volume. Direct observations of inhomogeneous deformations with a fracture zone after initial elastic strain were demonstrated for the thixotropic (“self-healing”) protein gel in [
42]. Destruction can be accompanied by sliding along the solid boundary surface of the experimental cell.
The strength of gels in the extension was also investigated, and many publications are devoted to this method. A detailed review [
44] considers various aspects of gel fracture, including its theoretical substantiation, and contains a large number of references to original publications.
2.3. Yielding Materials (Liquids)
The viscoelastic properties of a matter in a gel-like state, where the network is formed by transient bonds, depend on its structure. The direct experimental proof of a solid-like behavior of the gel-like state is the independence of the storage modulus of frequency accompanied by relatively low mechanical losses. There are numerous examples [
46,
47,
48,
49,
50,
51,
52] of such behavior, many of which are analyzed in reviews and monographs [
53,
54,
55]. The evolution of viscoelastic properties in the transition from yielding liquid to real gel is carefully described in [
56].
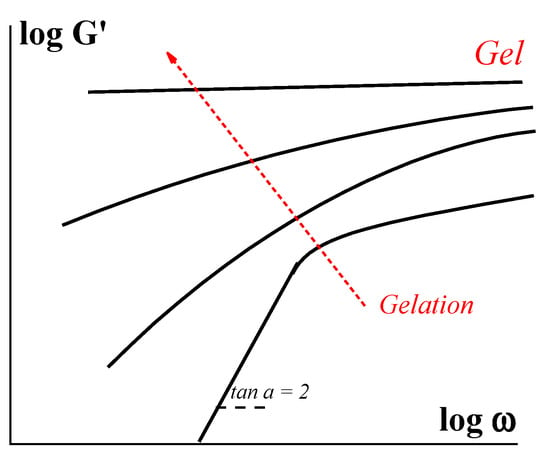
Developing viscoelasticity can be followed in parallel with the scheme in Figure 1. Initially, scholars deal with a usual Newtonian liquid with viscosity η0. Its loss modulus G″ is equal to (ωη0) in a whole frequency (ω) range and elasticity is negligible (G′ = 0). When a structural network appears, this is reflected in the appearance of non-Newtonian behavior, and the evolution of the storage modules occurs as schematically shown in Figure 2.
Figure 2. Evolution of the storage modulus in the process of gelation (scheme).
At the beginning of the process, a relaxation of the structuring liquid is close to a simple Maxwellian model, and the slope of the G′ (ω) dependence is equal to 2, which corresponds to a single relaxation time θ. Along with the development of the network, relaxation properties are characterized by a set of relaxation times that is reflected in a decrease of the slope of the G′ (ω) dependences. Finally, after reaching the gel state, scholars deal with a solid-like matter with the elastic modulus independent of frequency (upper straight line in Figure 2) as observed for any solid body.
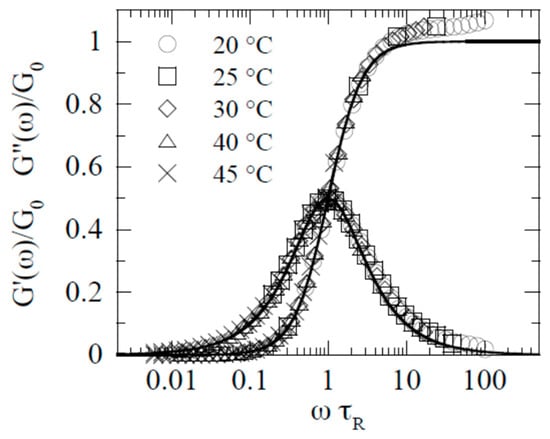
The width of the relaxation time depends on the nature of the matter. An extreme case is micellar solutions, which are yielding liquids. According to the review [
57], the viscoelastic properties of wormlike micelles are well-fitted by the Maxwell single-relaxation time model in a wide frequency range. This is illustrated in
Figure 3, where the solid lines are built strictly according to the Maxwell equation, and
τR and
G0 are relaxation time and the elastic modulus of the Maxwell model, respectively.
Figure 3. Viscoelastic properties of wormlike micelles (from [
57] with permission).
Such behavior in the linear region of viscoelasticity is quite typical of various micellar structures [
58,
59] and lamellar gels [
60]. The constants characterizing the relaxation properties of such soft matters are related to the classical relationship.
If a structural network is formed by macromolecular chains, the relaxation spectrum of the soft matter can be quite wide since the polymeric chains have many modes of relaxation (see, for example, [
61,
62]).
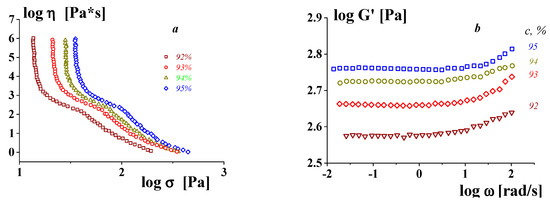
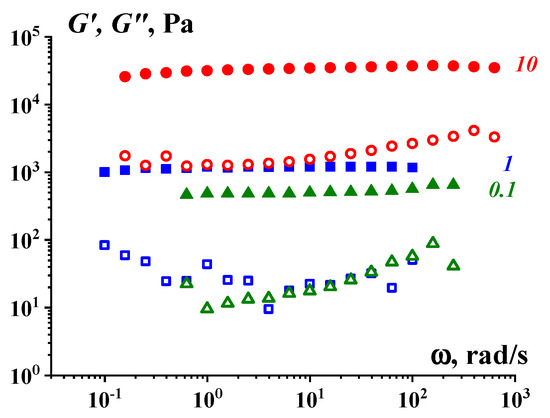
The concept of a gel-like state relates to different substances, including those that are not usually considered to be “gels”. The viscoelastic properties of highly concentrated emulsions presented in
Figure 4 are a typical example confirming this approach. One can see the correlation between the rheology of typical yielding materials (presented by the flow curves) and the frequency independence of the storage modulus in the gel-like state (at low stresses) [
63].
Figure 4. Flow curves–dependences of the apparent viscosity on shear stress (a) and frequency dependences of the storage modulus (b) in the gel-like state of low stresses for concentrated emulsions (these objects are liquid explosives.
Figure 5 demonstrates the other typical feature of the viscoelastic properties of a gel-like state of soft matter, where an intermolecular network in the polymer solution is created by silica nanoparticles. These have much higher values of the storage modulus in comparison with the loss modulus [
64].
Figure 5. Viscoelastic properties of the aqueous solutions of poly(ethylene oxide) with different concentrations (shown in the curves) in the presence of 3 vol.% of SiO
2. Filled symbols–storage modulus G′; open symbols–loss modulus, G″.
3. On the Yield Stress in Soft Matter
Measuring the yield stress, σ
Y, of viscopastic media is not such a simple task as it seems at first glance [
88,
89]. First of all, it is necessary to determine what is meant in a particular case by this value, i.e., an unambiguous definition of what scholars want to measure should be given. A milestone in this problem was the classic work of Bingham [
90]. He clearly distinguished two possible states of multicomponent multiphase materials: a solid (or gel-like) state, where the material exhibits only elastic deformations, and a liquid state, where this material can flow under the action of applied stresses. The boundary between these two states is the yield stress (or yield point) σ
Y, and its value was considered as a physical parameter of a substance.
Such an approach can often be found in modern publications on the characterization and comparison of various substances since it adequately describes the behavior of various concentrated suspensions, for example, bentonite colloidal systems [
91,
92], electrorheological liquids [
93], and supramolecular solutions [
94], as well as colloidal gels [
95,
96,
97], gels for 3D printing [
98], liquid crystal systems [
99], ferrofluids [
100,
101], and magnetorheological fluids [
102]. Yielding behavior and flow are important in many operations within the oil and gas industry [
103].
A visual analysis of many model situations using the Bingham understanding of yield stress is conducted in one review [
104]. Also, the Bingham model and its non-linear generalization are the basis for solving a lot of dynamic (boundary) problems. The known analytical solutions can be used in technological practice, for example, in designing the transport of cement mortars.
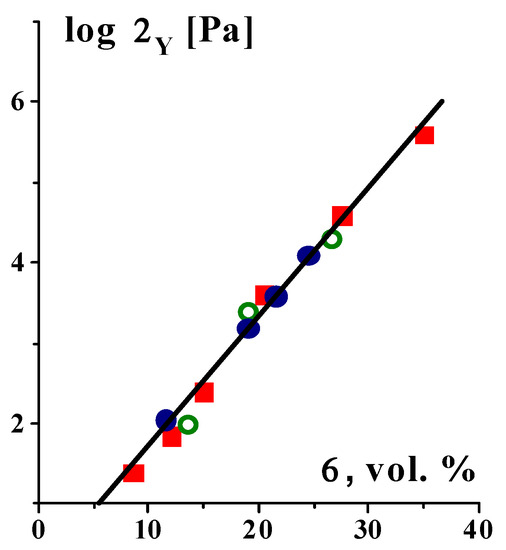
The yield stress value reflects the strength of a structure, which is created by intermolecular interactions of various types. The upper limit of the yield stress depends both on the nature of the bonds and on the concentration of the structure-forming components. The strength of the structure in highly filled compositions can exceed 10
5 Pa, and this is the structure formed specifically by the filler since the yield stress does not depend on the nature of the liquid matrix [
105] (see the example in
Figure 6).
Figure 6. Concentration dependence of the yield stress for dispersions of carbon black (surface 300 m
2/g) in different matrix–poly(butadiene)s with M
w = 1.35 × 10
5 and 1 × 10
4 Da and low-viscosity silicon oil–marked with various symbols; with various symbols calculated by the Casson and the Herschel–Bulkley models [
106], and by G′ (σ
0) curves. Original data.
However, this is true only for components that do not interact with each other. In fact, the matrix can strongly influence the structure formation in gels. Of course, other options are possible, in which the matrix will take part in the formation of the structure [
107]. An illustrative example of the formation of a continuous structure by silica in a polymer matrix depending on the pH of the medium is presented in [
108]. The formation of a three-dimensional structure has been shown in systems based on proteins with polysaccharides, the properties of which (
Figure 7) are determined by the composition of biopolymer supramolecular complexes [
48,
66].
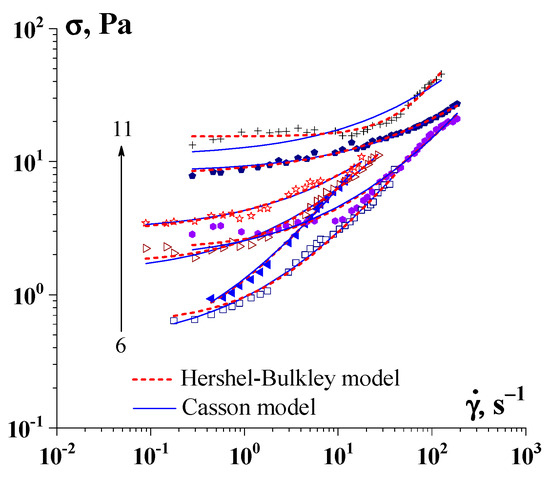
Figure 7. Determination of the yield stress by the Herschel–Bulkley model and the Casson model for hydrogel of gelatin-κ-carrageenan complexes (Cg = 1.0 wt.%). The change in the κ-carrageenan concentration is indicated by arrows, Ccar, wt.%: 6–0.100, 7–0.150, 8–0.175, 9–0.200, 10–0.400, 11–0.500.
The measurement of large values of the yield stress presents no fundamental difficulties. However, the issue of the minimum physically reasonable values of the yield stress is of special interest. In practice, using standard experimental techniques, it is not really possible to reliably measure the values of the yield stress below 1 Pa.
However, supramolecular structures have very low strength, and in this case, the values of the yield stress may be obtained by extrapolating experimental data. Such an approach sometimes gives minimum values of the yield stress in the range of 0.01–0.1 Pa at a concentration of the structure-forming phase significantly below 0.1% [
107,
109,
110]. Structure formation in organogels at extremely low concentrations of less than 10
–3% has been described [
111,
112].
The experimental measurement of the yield stress, σ
Y, should be based on a rigorous definition of this quantity. Indeed, after the question of whether there is a yield point, a counter-question follows: What is it? (that is, first of all, a definition of what the questioner understands by this value should be given). The concept advanced by Bingham seems quite clear [
90]. However, there are some theoretical and practical problems in determining the yield stress, even if we remain in the Bingham paradigm. Indeed, the yield point is defined by some equation (whether it be the Bingham, Herschel–Bulkley, Casson, or any other fitting equation; see
Figure 7) and extrapolation of the experimental data to the limit at
𝛾˙
→ 0.
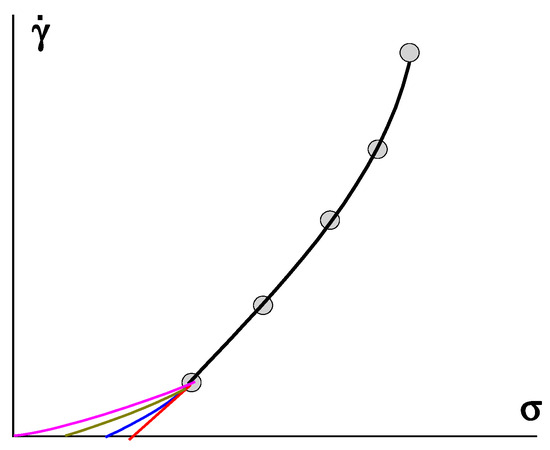
However, any extrapolation procedure leaves room for doubt, as illustrated in Figure 8, where different possible ways of extrapolating are shown. This procedure is especially dangerous when using a wide range of shear rates and presenting the experimental data in a log scale. It can be seen that it is possible to come to very different values of the yield stress (points of intersection of the lower curves with the σ-axis at 𝛾˙
= 0) and even the possible of the absence of a gel-like domain (σY = 0).
Figure 8. Various methods and results of extrapolation (number of lower curves) of experimental data–points and the fitting curve.
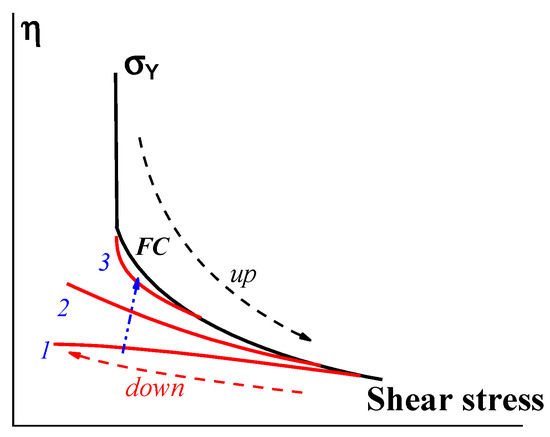
Bingham’s yield stress is a limiting case that may be true for compositions in which the dispersed component forms a rigid structure instantly breaking down at a certain stress, σ. However, even in these cases, the reverse restoration process can continue for a long time, and the different rates of approach to the initial yield stress (depending on the scanning speed) indicate the thixotropic nature of the substance (Figure 9).
Figure 9. Stress up-and-down scanning in measuring the flow curve. Curves 1, 2, and 3 correspond to a decreasing rate of scanning in decreasing shear stress (along the blue arrow).
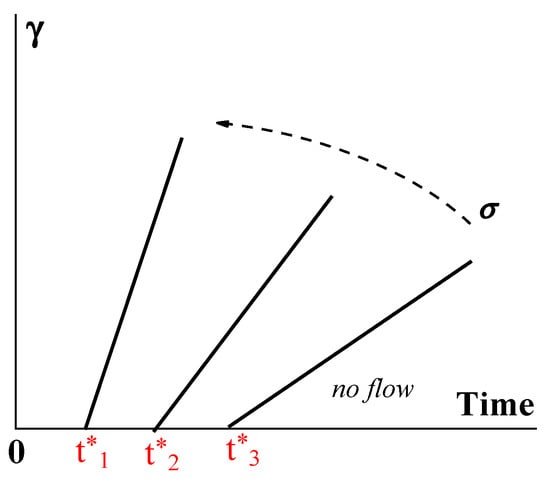
The gel-like structure can be different due to its physical origin, and consequently, its breaking up can happen in different ways. For example, instead of Bingham’s fragile jump-like transition from the gel-like to the liquid state, this transition can happen over time [
116,
117]. Then, the structure can be characterized by the lifetime t* decreasing (from t*
3 to t*
1) along with the increase in stress. Therefore, the flow does not begin immediately after the application of the load but only after some time, which characterizes the durability, or lifetime, of the structure (
Figure 10).
Figure 10. Developing deformation γ on time–Bingham yielding. The dotted arrows show the increase in shear stress.
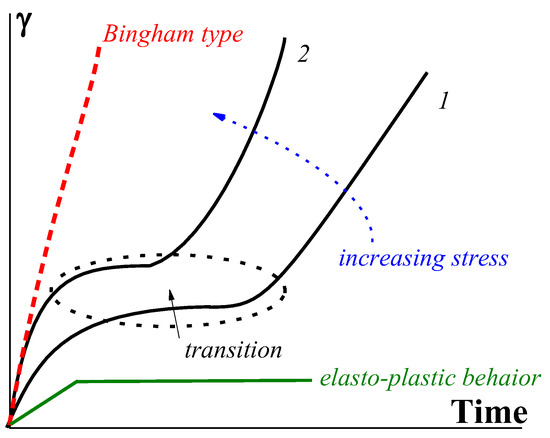
The function t* (σ) reflects the strength of a gel-like structure and thus replaces Bingham’s yield stress, σY. The slope of the straight lines in Figure 11 corresponds to the viscosity of a matter in a liquid state (above the gel-like-to-liquid transition).
Figure 11. Development of deformations in transition from the gel-like to plastic state (the stress for curve 2 is higher than that for curve 1).
The durability of rigid structures (in concentrated suspensions) is characterized by a rather strong dependence on stress. For example, the exponential-type
t* (
σ) dependence in yielding materials was proposed in [
118]:
where B and β are empirical constants in this equation. This equation is quite similar to Equation (1), fitting the strength of a material overall.
The time effects create a serious principal difficulty in finding the “true” yield point (which in reality may not exist), and the time effect leads to the conclusion that the yield stress should not be considered a material property since it depends on the prehistory of deformation and the rest of the sample [
119].
The rheological properties of yielding materials are similar to those of non-fluid gels at 𝜎<𝜎𝑌., where only small elastic deformations are possible. At 𝜎>𝜎𝑌, these elastic deformations are negligible in comparison to the irreversible deformation of flow, and therefore, they are not shown in Figure 10. However, it is reasonable to assume that the deformation during the initial stage of loading at σ > σY is not elastic but viscoelastic.
The dependences of such a type presented in [
120] can be reconstructed to give the
t* (σ) dependence, which reflects the durability of a solid-like structure. This is shown in
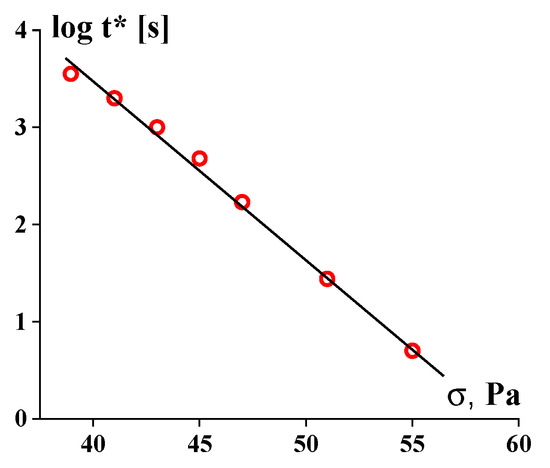
Figure 12 (based on the data from
Figure 3 in this publication).
Figure 12. Long-term shear-induced solid-to-liquid transition for 51% suspension of Kaolin.
These experimental data are approximated by the standard exponential dependence (5) with β = 0.414 Pa−1.
Therefore, two limiting types of rheological behavior should be distinguished: Bingham’s one with an immediate increase in deformation at 𝜎>𝜎𝑌
and viscoelastic behavior at
σ <
σY (for soft matter). The shear-induced transition from initial viscoelastic deformations to flow might not be sharp, as shown in
Figure 13, but smooth.
4. Sol–Gel Transition
The sol–gel transition is represented by the diagram in Figure 1. This already suggests that such a transition occurs over time, although it can also be considered as a transition through the yield point since, according to the physical meaning, this is a transition from a liquid state to a solid-like state.
As emphasized above, it is necessary to distinguish between a sol-to-gel transition to a solid gel state and a sol-to-gel-like transition to a yielding material (liquid), although the term “gelation” is used for both processes.
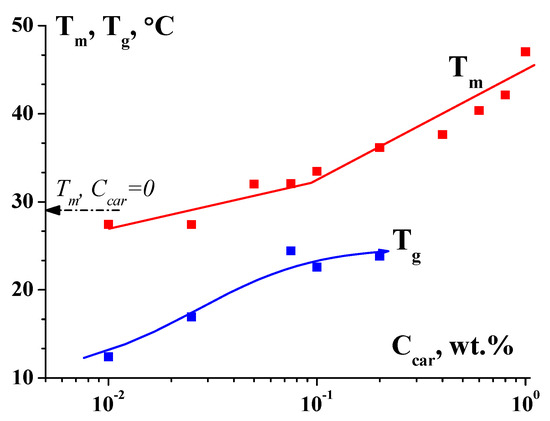
Traditionally, people working with weak organic and especially bioorganic compounds prefer to talk about sol–gel (or gel–sol) transitions, while those who work with coarser multicomponent materials interpret them as yielding. Meanwhile, in both cases, the physical content is the same—the formation of a network of the transient bonds of various types in a liquid. Moreover, the situation is quite common when the same composition, depending on the temperature, can form a true gel (below the glass transition temperature, T
g). And at higher temperatures (above the melting temperature, T
m), the composition becomes viscous liquid. In the temperature range T
g–T
m, this is a yielding material (
Figure 14) [
66].
Figure 14. Dependence of melting (Tm) and gelling (Tg) temperatures on the κ-carrageenan-to-gelatin mass ratio Z in mixed hydrogels; gelatin concentration CG = 1.0 wt.%. Original data.
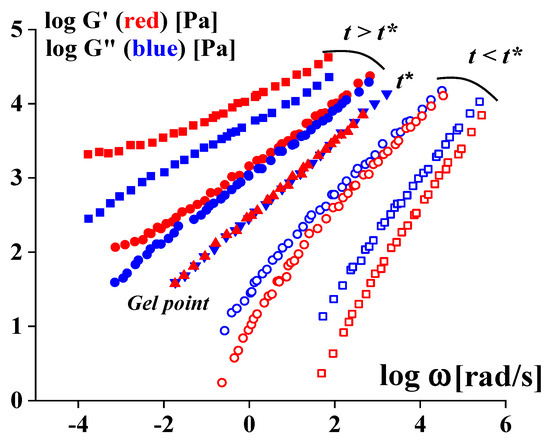
It is quite evident that gelation leads to the transformation of the viscoelastic properties of the matter. This basic concept was developed based on experiments with stoichiometric balanced cross-linking PDMS [
175,
176]. It was shown that the gel point is characterized by the equality of G′ (ω) = G″ (ω) over a very wide frequency range (but not at a single point, as assumed sometimes). In this threshold state, the following simple law holds:
where K is a temperature-sensitive factor.
Figure 15 shows how viscoelastic properties of the liquid before gelation (at t < t*) pass into properties of a typical gel (at t > t*), and this is accompanied by a change in the ratio of G′ and G″: in a liquid state, G″ > G′, and in a gel (or gel-like) state, G″ < G′.
Figure 15. Experimental data illustrating an evolution of viscoelastic properties at the liquid-to-gel transition, according to a concept formulated in [
176].
G′ (
ω) and
G″ (
ω) are used in the reduced form, and the constants are omitted; every pair of curves corresponds to the different moments of the gelation.
In addition, it was shown that at the gel point, a very simple relationship exists for the relaxation modulus,
Gr =
S·t−1/2, and only a single parameter
S, referred to as the “strength” of a gel network, is enough to characterize the gel properties. The development of this approach for stoichiometric imbalanced cross-linking was discussed in [
177]. Initially, this approach to determining the gel point was developed for chemical gels. Later, it was shown that the same method can be applied to sol–gel transition for gels formed by transient bonds [
178].
The sol–gel transition is a rather special state of the system associated with the formation of the space percolation network. In the publications of Winter et al. cited above, as well as in the review of earlier studies [
182], it was shown that the rheological behavior in the vicinity of this critical point is characterized by power law dependences of viscoelastic functions on frequency or time. These power-law dependences were treated as a consequence of the fractal structure of the matter near the gel point [
183]. It was also shown that the approach to the gel point is accompanied by the formation of space clusters in the pre-gel state [
184].
Both the agglomeration and the deagglomeration (breakdown) of the particle network in polymer nanocomposites are affected by the shear flow, resulting in shear-induced liquid-solid (sol–gel) transition and shear-induced solid-liquid (gel–sol) transition, respectively. It was shown [
186] that the percolation threshold of both transitions under shear-induced agglomeration and breakdown processes depends on the shear rate under a steady shear condition.
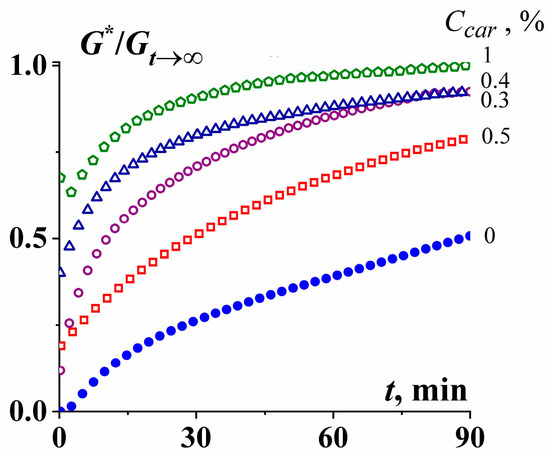
Figure 16 demonstrates that the addition of κ-carrageenan to gelatin solution increases the rate of gelation in the isothermal conditions as followed by changes in the time dependence of the complex elastic modulus,
G* (
t), for samples with different κ-carrageenan content [
153].
Figure 16. Kinetics of gelation as followed by an increase of the dynamic modulus G* normalized by its limiting value, G*t→∞, at CG = 2 g/100 g and different concentrations of κ-carrageenan.
The temperature and frequency dependences of rheological parameters can sometimes be considered interchangeable. This is an attempt to apply a similar method to the temperature-frequency equivalence in the linear viscoelasticity of polymers. In the case of viscoelasticity, this principle only works if the shape of the relaxation spectrum (as a reflection of the internal structure of the substance) does not change. In the case of gelation, both an increase in temperature and a reduction in the frequency helps to accelerate the gel-to-sol transition (“melting” of the gel).
In technological practice, quite simple “visual” methods of the sol-to-gel transition are widely used. They are based on the standardized estimation of the moment of the loss of fluidity. Some simple proposed methods for finding the yield transition are based on elongation deformation considering the drop formation during the extrusion of a yield stress fluid in the air [
87,
199,
200]. Observation of the “falling ball”, displacement of the meniscus, the capillary method [
201,
202], the inclined plate method [
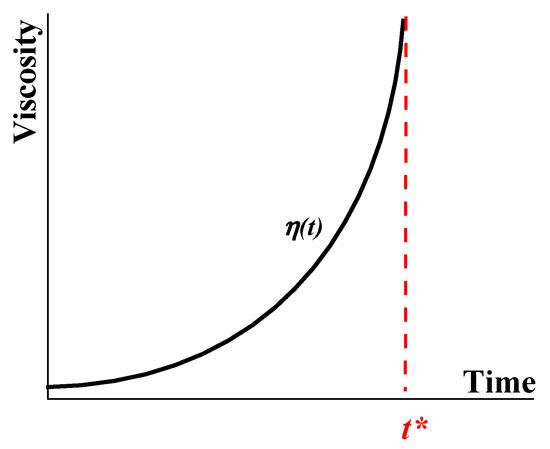
203], and determination of the cloud point of crude oils, or the temperature of wax appearance (WAT), according to ASTM D-5853, do not provide the necessary accuracy and reliability in measuring the gelation time, although they can be useful for preliminary characterization of the samples. Another simplified method for estimating the sol–gel point can be used based on measurements of the time dependence of the apparent viscosity at constant shear stress. This dependence is shown in
Figure 17.
Figure 17. Typical evolution of viscosity over tine in the gelation process up to the gel point
t*.
5. Conclusions
It is necessary to distinguish solid gels and yielding liquids, which can form a solid-like (gel-like) structure that is destroyed by increasing stress. The proposed rigorous definition of gels as solid materials contradicts the habitual treating of colloidal yielding substances as “gels” but advances a general understanding of the rheological behavior of all materials demonstrating yield stress (not correctly called viscoplastic media).
The main peculiarity of various soft multicomponent subjects is the ability to transit from a solid-like to a liquid state due to incomplete phase decomposition. This leads to the formation of a structured network with some strength, in which a significant amount of solvent is immobilized. The transition between these states—also sometimes called the “gel-sol transition”—can be caused by external thermodynamic factors, which are typical for solid-like gels (temperature, changes in the environment, etc.) but can also occur under the action of applied mechanical forces. It is the latter case that is inherent in a large number of so-called “yielding materials” (liquids), and the solid-to-liquid transition takes place at some threshold, certified as yield stress. This point is quite clearly determined for rigid and fragile structure networks, but in the vast majority of real materials (such as lubricants, paints, foodstuffs, clay, soils, biological hydrogels, and natural phenomena such as mudflows), this transition can be extended over time. The time effects of such objects are similar to the durability of a solid-like structure and its deformation-induced thixotropy. This leads to practical uncertainty in estimating the solid-to-liquid transition point.
This entry is adapted from the peer-reviewed paper 10.3390/gels9090715