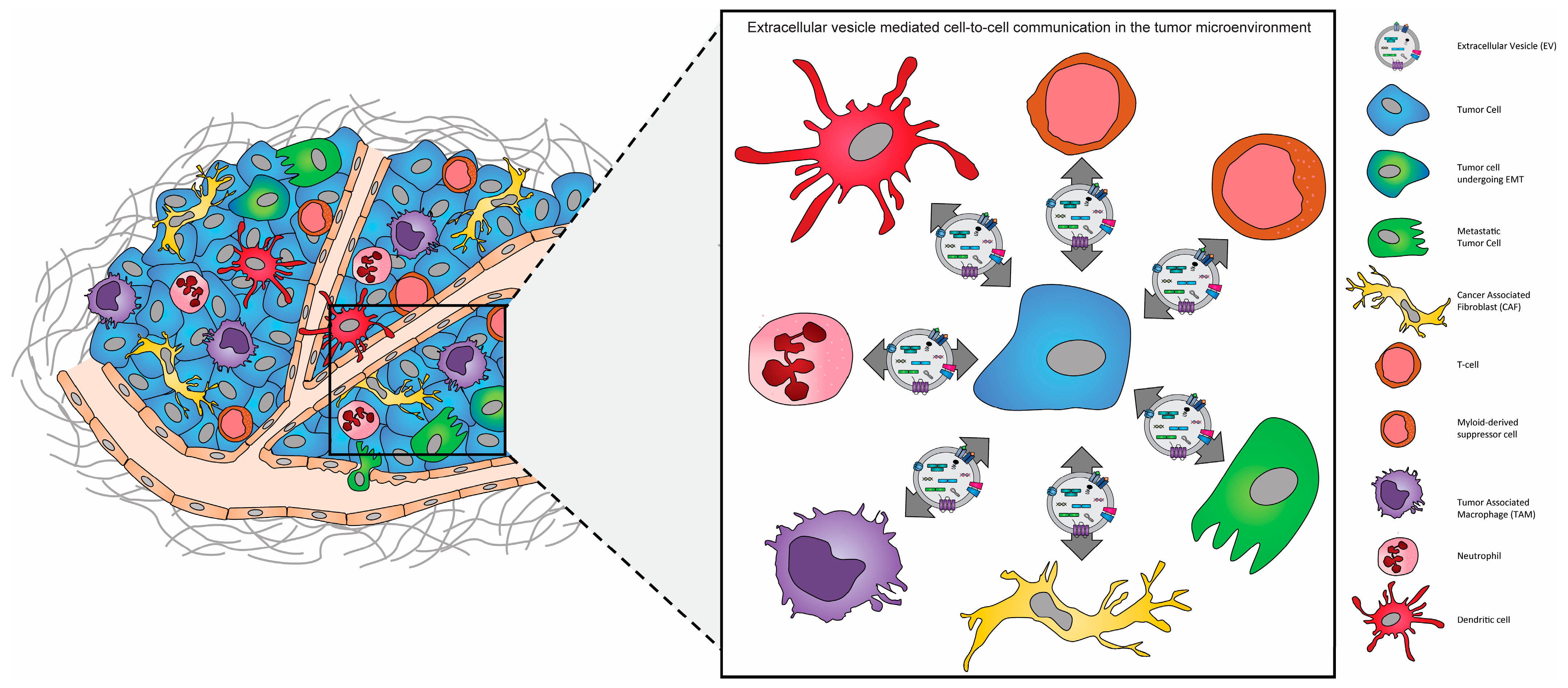
It is increasingly recognized that the complex heterogeneous nature of the tumor microenvironment plays a critical role in the evolution and progression of cancer. The communication between matrix cells within the tumor microenvironment (TME), via extracellular vesicles, serves as an essential mediator for the development, recurrence, and metastatic dissemination of several cancer types. The role of extracellular vesicles (EVs) within the tumor microenvironment appears to be multifaceted and bidirectional as the initial release of EVs from cancer cells leads to the recruitment and activation of stromal cells, which includes cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAM), cancer-associated endothelial cells (CAEC), and mesenchymal stem cells (MSC). As tumor growth progresses, evidence suggests that EVs released from surrounding stromal cells drive the epithelial-to-mesenchymal transition (EMT) of cancer cells, and thus their progression to more metastatic phenotypes. Complementarily, as the release of EVs is enhanced during tumor growth, for certain tumors it allows their widespread diffusion and provides them with the potential to establish the pre-metastatic niche, which is necessary for the successful dissemination, colonization, and expansion of these cells to distant organ sites.
- extracellular vesicles
- cancer
- metastasis
- biomarkers
- cell-to-cell communication
1. Cancer-Associated Fibroblasts (CAFs)
2. Tumor-Associated Macrophages (TAMs)
3. Tumor Endothelial Cells (TECs)
4. Tumor-Infiltrating Lymphocytes (TILs)
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines11092534
References
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model. Mech. 2018, 11, dmm029447.
- Manoukian, P.; Bijlsma, M.; van Laarhoven, H. The Cellular Origins of Cancer-Associated Fibroblasts and Their Opposing Contributions to Pancreatic Cancer Growth. Front. Cell Dev. Biol. 2021, 9, 743907.
- Wright, K.; Ly, T.; Kriet, M.; Czirok, A.; Thomas, S.M. Cancer-Associated Fibroblasts: Master Tumor Microenvironment Modifiers. Cancers 2023, 15, 1899.
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186.
- Mitchell, M.I.; Engelbrecht, A.-M. Metabolic hijacking: A survival strategy cancer cells exploit? Crit. Rev. Oncol. Hematol. 2017, 109, 1–8.
- Dvorak, H.F. Tumors: Wounds That Do Not Heal—Redux. Cancer Immunol. Res. 2015, 3, 1–11.
- Hua, Y.; Bergers, G. Tumors vs. Chronic Wounds: An Immune Cell's Perspective. Front. Immunol. 2019, 10, 2178.
- Deyell, M.; Garris, C.S.; Laughney, A.M. Cancer metastasis as a non-healing wound. Br. J. Cancer 2021, 124, 1491–1502.
- Öhlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523.
- Ziani, L.; Chouaib, S.; Thiery, J. Alteration of the Antitumor Immune Response by Cancer-Associated Fibroblasts. Front. Immunol. 2018, 9, 414.
- Li, C.; Teixeira, A.F.; Zhu, H.-J.; Dijke, P.T. Cancer associated-fibroblast-derived exosomes in cancer progression. Mol. Cancer 2021, 20, 154.
- Peng, Z.; Tong, Z.; Ren, Z.; Ye, M.; Hu, K. Cancer-associated fibroblasts and its derived exosomes: A new perspective for reshaping the tumor microenvironment. Mol. Med. 2023, 29, 66.
- Gu, J.; Qian, H.; Shen, L.; Zhang, X.; Zhu, W.; Huang, L.; Yan, Y.; Mao, F.; Zhao, C.; Shi, Y.; et al. Gastric Cancer Exosomes Trigger Differentiation of Umbilical Cord Derived Mesenchymal Stem Cells to Carcinoma-Associated Fibroblasts through TGF-β/Smad Pathway. PLoS ONE 2012, 7, e52465.
- Webber, J.; Steadman, R.; Mason, M.D.; Tabi, Z.; Clayton, A. Cancer Exosomes Trigger Fibroblast to Myofibroblast Differentiation. Cancer Res. 2010, 70, 9621–9630.
- Cho, J.A.; Park, H.; Lim, E.H.; Kim, K.H.; Choi, J.S.; Lee, J.H.; Shin, J.W.; Lee, K.W. Exosomes from ovarian cancer cells induce adipose tissue-derived mesenchymal stem cells to acquire the physical and functional characteristics of tumor-supporting myofibroblasts. Gynecol. Oncol. 2011, 123, 379–386.
- Cho, J.A.; Park, H.; Lim, E.H.; Lee, K.W. Exosomes from breast cancer cells can convert adipose tissue-derived mesenchymal stem cells into myofibroblast-like cells. Int. J. Oncol. 2011, 40, 130–138.
- Catteau, X.; Simon, P.; Noёl, J.-C. Stromal expression of matrix metalloproteinase 2 in cancer-associated fibrobasts is strongly related to human epidermal growth factor receptor 2 status in invasive breast carcinoma. Mol. Clin. Oncol. 2016, 4, 375–378.
- Luga, V.; Wrana, J.L. Tumor–Stroma Interaction: Revealing Fibroblast-Secreted Exosomes as Potent Regulators of Wnt-Planar Cell Polarity Signaling in Cancer Metastasis. Cancer Res. 2013, 73, 6843–6847.
- Otranto, M.; Sarrazy, V.; Bonté, F.; Hinz, B.; Gabbiani, G.; Desmouliere, A. The role of the myofibroblast in tumor stroma remodeling. Cell Adhes. Migr. 2012, 6, 203–219.
- Luga, V.; Zhang, L.; Viloria-Petit, A.M.; Ogunjimi, A.A.; Inanlou, M.R.; Chiu, E.; Buchanan, M.; Hosein, A.N.; Basik, M.; Wrana, J.L. Exosomes Mediate Stromal Mobilization of Autocrine Wnt-PCP Signaling in Breast Cancer Cell Migration. Cell 2012, 151, 1542–1556.
- Sidhu, S.S.; Mengistab, A.T.; Tauscher, A.N.; LaVail, J.; Basbaum, C. The microvesicle as a vehicle for EMMPRIN in tumor–stromal interactions. Oncogene 2004, 23, 956–963.
- Ramteke, A.; Ting, H.; Agarwal, C.; Mateen, S.; Somasagara, R.; Hussain, A.; Graner, M.; Frederick, B.; Agarwal, R.; Deep, G. Exosomes secreted under hypoxia enhance invasiveness and stemness of prostate cancer cells by targeting adherens junction molecules. Mol. Carcinog. 2013, 54, 554–565.
- Krzyszczyk, P.; Schloss, R.; Palmer, A.; Berthiaume, F. The Role of Macrophages in Acute and Chronic Wound Healing and Interventions to Promote Pro-wound Healing Phenotypes. Front. Physiol. 2018, 9, 419.
- Logozzi, M.; Mizzoni, D.; Capasso, C.; Del Prete, S.; Di Raimo, R.; Falchi, M.; Angelini, D.F.; Sciarra, A.; Maggi, M.; Supuran, C.T.; et al. Plasmatic exosomes from prostate cancer patients show increased carbonic anhydrase IX expression and activity and low pH. J. Enzym. Inhib. Med. Chem. 2019, 35, 280–288.
- Henze, A.-T.; Mazzone, M. The impact of hypoxia on tumor-associated macrophages. J. Clin. Investig. 2016, 126, 3672–3679.
- Pollard, J.W. Macrophages define the invasive microenvironment in breast cancer. J. Leukoc. Biol. 2008, 84, 623–630.
- Grivennikov, S.I.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.Y.; Osterreicher, C.H.; Hung, K.E.; et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012, 491, 254–258.
- Kong, L.; Zhou, Y.; Bu, H.; Lv, T.; Shi, Y.; Yang, J. Deletion of interleukin-6 in monocytes/macrophages suppresses the initiation of hepatocellular carcinoma in mice. J. Exp. Clin. Cancer Res. 2016, 35, 131.
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896.
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS–) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084, Erratum in Front. Immunol. 2020, 11, 234.
- Mould, K.J.; Jackson, N.D.; Henson, P.M.; Seibold, M.; Janssen, W.J. Single cell RNA sequencing identifies unique inflammatory airspace macrophage subsets. JCI Insight 2019, 4, e126556.
- Qian, B.-Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51.
- Chen, Q.; Li, Y.; Gao, W.; Chen, L.; Xu, W.; Zhu, X. Exosome-Mediated Crosstalk Between Tumor and Tumor-Associated Macrophages. Front. Mol. Biosci. 2021, 8, 764222.
- Seo, N.; Shirakura, Y.; Tahara, Y.; Momose, F.; Harada, N.; Ikeda, H.; Akiyoshi, K.; Shiku, H. Activated CD8+ T cell extracellular vesicles prevent tumour progression by targeting of lesional mesenchymal cells. Nat. Commun. 2018, 9, 435.
- Baig, M.S.; Roy, A.; Rajpoot, S.; Liu, D.; Savai, R.; Banerjee, S.; Kawada, M.; Faisal, S.M.; Saluja, R.; Saqib, U.; et al. Tumor-derived exosomes in the regulation of macrophage polarization. Inflamm. Res. 2020, 69, 435–451.
- Marleau, A.M.; Chen, C.-S.; Joyce, J.A.; Tullis, R.H. Exosome removal as a therapeutic adjuvant in cancer. J. Transl. Med. 2012, 10, 134.
- LeBleu, V.S.; Kalluri, R. Exosomes Exercise Inhibition of Anti-Tumor Immunity during Chemotherapy. Immunity 2019, 50, 547–549.
- Yang, Q.; Xu, J.; Gu, J.; Shi, H.; Zhang, J.; Zhang, J.; Chen, Z.; Fang, X.; Zhu, T.; Zhang, X. Extracellular Vesicles in Cancer Drug Resistance: Roles, Mechanisms, and Implications. Adv. Sci. 2022, 9, e2201609.
- Zhou, X.; Liu, Q.; Wang, X.; Yao, X.; Zhang, B.; Wu, J.; Sun, C. Exosomal ncRNAs facilitate interactive ‘dialogue’ between tumor cells and tumor-associated macrophages. Cancer Lett. 2022, 552, 215975.
- Zhou, J.; Li, X.; Wu, X.; Zhang, T.; Zhu, Q.; Wang, X.; Wang, H.; Wang, K.; Lin, Y.; Wang, X. Exosomes Released from Tumor-Associated Macrophages Transfer miRNAs That Induce a Treg/Th17 Cell Imbalance in Epithelial Ovarian Cancer. Cancer Immunol. Res. 2018, 6, 1578–1592.
- Yin, K.; Wang, S.; Zhao, R.C. Exosomes from mesenchymal stem/stromal cells: A new therapeutic paradigm. Biomark. Res. 2019, 7, 8.
- Zhao, S.; Mi, Y.; Guan, B.; Zheng, B.; Wei, P.; Gu, Y.; Zhang, Z.; Cai, S.; Xu, Y.; Li, X.; et al. Tumor-derived exosomal miR-934 induces macrophage M2 polarization to promote liver metastasis of colorectal cancer. J. Hematol. Oncol. 2020, 13, 156, Erratum in J. Hematol. Oncol. 2021, 14, 33.
- Takano, Y.; Masuda, T.; Iinuma, H.; Yamaguchi, R.; Sato, K.; Tobo, T.; Hirata, H.; Kuroda, Y.; Nambara, S.; Hayashi, N.; et al. Circulating exosomal microRNA-203 is associated with metastasis possibly via inducing tumor-associated macrophages in colorectal cancer. Oncotarget 2017, 8, 78598–78613.
- Shinohara, H.; Kuranaga, Y.; Kumazaki, M.; Sugito, N.; Yoshikawa, Y.; Takai, T.; Taniguchi, K.; Ito, Y.; Akao, Y. Regulated Polarization of Tumor-Associated Macrophages by miR-145 via Colorectal Cancer–Derived Extracellular Vesicles. J. Immunol. 2017, 199, 1505–1515.
- Cai, J.; Qiao, B.; Gao, N.; Lin, N.; He, W. Oral squamous cell carcinoma-derived exosomes promote M2 subtype macrophage polarization mediated by exosome-enclosed miR-29a-3p. Am. J. Physiol. Cell. Physiol. 2019, 316, C731–C740.
- Chen, X.; Ying, X.; Wang, X.; Wu, X.; Zhu, Q.; Wang, X. Exosomes derived from hypoxic epithelial ovarian cancer deliver microRNA-940 to induce macrophage M2 polarization. Oncol. Rep. 2017, 38, 522–528.
- Chen, X.; Zhou, J.; Li, X.; Wang, X.; Lin, Y.; Wang, X. Exosomes derived from hypoxic epithelial ovarian cancer cells deliver microRNAs to macrophages and elicit a tumor-promoted phenotype. Cancer Lett. 2018, 435, 80–91.
- Wang, X.; Luo, G.; Zhang, K.; Cao, J.; Huang, C.; Jiang, T.; Liu, B.; Su, L.; Qiu, Z. Hypoxic Tumor-Derived Exosomal miR-301a Mediates M2 Macrophage Polarization via PTEN/PI3Kγ to Promote Pancreatic Cancer Metastasis. Cancer Res. 2018, 78, 4586–4598.
- McDonald, D.M.; Choyke, P.L. Imaging of angiogenesis: From microscope to clinic. Nat. Med. 2003, 9, 713–725.
- di Tomaso, E.; Capen, D.; Haskell, A.; Hart, J.; Logie, J.J.; Jain, R.K.; McDonald, D.M.; Jones, R.; Munn, L.L. Mosaic Tumor Vessels: Cellular Basis and Ultrastructure of Focal Regions Lacking Endothelial Cell Markers. Cancer Res. 2005, 65, 5740–5749.
- McDonald, D.M.; Baluk, P. Significance of blood vessel leakiness in cancer. Cancer Res. 2002, 62, 5381–5385.
- Boucher, Y.; Jain, R.K. Microvascular pressure is the principal driving force for interstitial hypertension in solid tumors: Implications for vascular collapse. Cancer Res. 1992, 52, 5110–5114.
- Seaman, S.; Stevens, J.; Yang, M.Y.; Logsdon, D.; Graff-Cherry, C.; Croix, B.S. Genes that Distinguish Physiological and Pathological Angiogenesis. Cancer Cell 2007, 11, 539–554.
- Goh, P.P.; Sze, D.M.; Roufogalis, B.D. Molecular and Cellular Regulators of Cancer Angiogenesis. Curr. Cancer Drug Targets 2007, 7, 743–758.
- Hida, K.; Klagsbrun, M. A New Perspective on Tumor Endothelial Cells: Unexpected Chromosome and Centrosome Abnormalities. Cancer Res. 2005, 65, 2507–2510.
- Hida, K.; Maishi, N.; Annan, D.A.; Hida, Y. Contribution of Tumor Endothelial Cells in Cancer Progression. Int. J. Mol. Sci. 2018, 19, 1272.
- Amin, D.N.; Hida, K.; Bielenberg, D.R.; Klagsbrun, M. Tumor Endothelial Cells Express Epidermal Growth Factor Receptor (EGFR) but not ErbB3 and Are Responsive to EGF and to EGFR Kinase Inhibitors. Cancer Res. 2006, 66, 2173–2180.
- Tsuchiya, K.; Hida, K.; Hida, Y.; Muraki, C.; Ohga, N.; Akino, T.; Kondo, T.; Miseki, T.; Nakagawa, K.; Shindoh, M.; et al. Adrenomedullin antagonist suppresses tumor formation in renal cell carcinoma through inhibitory effects on tumor endothelial cells and endothelial progenitor mobilization. Int. J. Oncol. 2010, 36, 1379–1386.
- Matsuda, K.; Ohga, N.; Hida, Y.; Muraki, C.; Tsuchiya, K.; Kurosu, T.; Akino, T.; Shih, S.-C.; Totsuka, Y.; Klagsbrun, M.; et al. Isolated tumor endothelial cells maintain specific character during long-term culture. Biochem. Biophys. Res. Commun. 2010, 394, 947–954.
- Heldin, C.-H.; Rubin, K.; Pietras, K.; Östman, A. High interstitial fluid pressure—An obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813.
- Butler, J.M.; Kobayashi, H.; Rafii, S. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat. Rev. Cancer 2010, 10, 138–146.
- Cao, Z.; Ding, B.-S.; Guo, P.; Lee, S.B.; Butler, J.M.; Casey, S.C.; Simons, M.; Tam, W.; Felsher, D.W.; Shido, K.; et al. Angiocrine Factors Deployed by Tumor Vascular Niche Induce B Cell Lymphoma Invasiveness and Chemoresistance. Cancer Cell 2014, 25, 350–365.
- Cao, Z.; Scandura, J.M.; Inghirami, G.G.; Shido, K.; Ding, B.-S.; Rafii, S. Molecular Checkpoint Decisions Made by Subverted Vascular Niche Transform Indolent Tumor Cells into Chemoresistant Cancer Stem Cells. Cancer Cell 2016, 31, 110–126.
- Mao, Y.; Wang, Y.; Dong, L.; Zhang, Y.; Zhang, Y.; Wang, C.; Zhang, Q.; Yang, S.; Cao, L.; Zhang, X.; et al. Hypoxic exosomes facilitate angiogenesis and metastasis in esophageal squamous cell carcinoma through altering the phenotype and transcriptome of endothelial cells. J. Exp. Clin. Cancer Res. 2019, 38, 389.
- Chen, C.; Liu, Y.; Liu, L.; Si, C.; Xu, Y.; Wu, X.; Wang, C.; Sun, Z.; Kang, Q. Exosomal circTUBGCP4 promotes vascular endothelial cell tipping and colorectal cancer metastasis by activating Akt signaling pathway. J. Exp. Clin. Cancer Res. 2023, 42, 46.
- Biagioni, A.; Laurenzana, A.; Menicacci, B.; Peppicelli, S.; Andreucci, E.; Bianchini, F.; Guasti, D.; Paoli, P.; Serratì, S.; Mocali, A.; et al. uPAR-expressing melanoma exosomes promote angiogenesis by VE-Cadherin, EGFR and uPAR overexpression and rise of ERK1,2 signaling in endothelial cells. Cell. Mol. Life Sci. 2021, 78, 3057–3072.
- Zeng, Z.; Li, Y.; Pan, Y.; Lan, X.; Song, F.; Sun, J.; Zhou, K.; Liu, X.; Ren, X.; Wang, F.; et al. Cancer-derived exosomal miR-25-3p promotes pre-metastatic niche formation by inducing vascular permeability and angiogenesis. Nat. Commun. 2018, 9, 5395.
- Pruneri, G.; Vingiani, A.; Denkert, C. Tumor infiltrating lymphocytes in early breast cancer. Breast 2017, 37, 207–214.
- Li, R.; Cao, L. The role of tumor-infiltrating lymphocytes in triple-negative breast cancer and the research progress of adoptive cell therapy. Front. Immunol. 2023, 14, 1194020.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Kitamura, T.; Qian, B.-Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86.
- Zhang, Z.; Liu, S.; Zhang, B.; Qiao, L.; Zhang, Y. T Cell Dysfunction and Exhaustion in Cancer. Front. Cell Dev. Biol. 2020, 8, 17.
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925.
- Whiteside, T.L. Tumor-Infiltrating Lymphocytes and Their Role in Solid Tumor Progression. Exp. Suppl. 2022, 113, 89–106.
- Dieci, M.V.; Mathieu, M.C.; Guarneri, V.; Conte, P.; Delaloge, S.; Andre, F.; Goubar, A. Prognostic and predictive value of tumor-infiltrating lymphocytes in two phase III randomized adjuvant breast cancer trials. Ann. Oncol. 2015, 26, 1698–1704.
- Zhao, Y.; Ge, X.; He, J.; Cheng, Y.; Wang, Z.; Wang, J.; Sun, L. The prognostic value of tumor-infiltrating lymphocytes in colorectal cancer differs by anatomical subsite: A systematic review and meta-analysis. World J. Surg. Oncol. 2019, 17, 85.
- Hashemi, S.; Fransen, M.; Niemeijer, A.; Ben Taleb, N.; Houda, I.; Veltman, J.; Commissaris, A.B.; Daniels, H.; Crombag, L.; Radonic, T.; et al. Surprising impact of stromal TIL’s on immunotherapy efficacy in a real-world lung cancer study. Lung Cancer 2021, 153, 81–89.
- Haj-Shomaly, J.; Vorontsova, A.; Barenholz-Cohen, T.; Levi-Galibov, O.; Devarasetty, M.; Timaner, M.; Raviv, Z.; Cooper, T.J.; Soker, S.; Hasson, P.; et al. T Cells Promote Metastasis by Regulating Extracellular Matrix Remodeling following Chemotherapy. Cancer Res. 2022, 82, 278–291.
- Hall, M.; Liu, H.; Malafa, M.; Centeno, B.; Hodul, P.J.; Pimiento, J.; Pilon-Thomas, S.; Sarnaik, A.A. Expansion of tumor-infiltrating lymphocytes (TIL) from human pancreatic tumors. J. Immunother. Cancer 2016, 4, 61.
- Baghaei, K.; Tokhanbigli, S.; Asadzadeh, H.; Nmaki, S.; Zali, M.R.; Hashemi, S.M. Exosomes as a novel cell-free therapeutic approach in gastrointestinal diseases. J. Cell. Physiol. 2019, 234, 9910–9926.
- Kishton, R.J.; Vodnala, S.K.; Vizcardo, R.; Restifo, N.P. Next generation immunotherapy: Enhancing stemness of polyclonal T cells to improve anti-tumor activity. Curr. Opin. Immunol. 2022, 74, 39–45.
- Olejarz, W.; Dominiak, A.; Żołnierzak, A.; Kubiak-Tomaszewska, G.; Lorenc, T. Tumor-Derived Exosomes in Immunosuppression and Immunotherapy. J. Immunol. Res. 2020, 2020, 6272498.
- Whiteside, T.L. Immune modulation of T-cell and NK (natural killer) cell activities by TEXs (tumour-derived exosomes). Biochem. Soc. Trans. 2013, 41, 245–251.
- Whiteside, T.L. Exosomes carrying immunoinhibitory proteins and their role in cancer. Clin. Exp. Immunol. 2017, 189, 259–267.
- Nakazawa, Y.; Nishiyama, N.; Koizumi, H.; Kanemaru, K.; Nakahashi-Oda, C.; Shibuya, A. Tumor-derived extracellular vesicles regulate tumor-infiltrating regulatory T cells via the inhibitory immunoreceptor CD300a. eLife 2021, 10, e61999.
- Wieckowski, E.U.; Visus, C.; Szajnik, M.; Szczepanski, M.J.; Storkus, W.J.; Whiteside, T.L. Tumor-Derived Microvesicles Promote Regulatory T Cell Expansion and Induce Apoptosis in Tumor-Reactive Activated CD8+ T Lymphocytes. J. Immunol. 2009, 183, 3720–3730.
- Wieckowski, E.; Whiteside, T.L. Human Tumor-Derived vs Dendritic Cell-Derived Exosomes Have Distinct Biologic Roles and Molecular Profiles. Immunol. Res. 2006, 36, 247–254.
- Montes, C.L.; Chapoval, A.I.; Nelson, J.; Orhue, V.; Zhang, X.; Schulze, D.H.; Strome, S.E.; Gastman, B.R. Tumor-Induced Senescent T Cells with Suppressor Function: A Potential Form of Tumor Immune Evasion. Cancer Res. 2008, 68, 870–879.
- Zhang, Y.; Pfannenstiel, L.W.; Bolesta, E.; Montes, C.L.; Zhang, X.; Chapoval, A.I.; Gartenhaus, R.B.; Strome, S.E.; Gastman, B.R. Interleukin-7 Inhibits Tumor-Induced CD27−CD28− Suppressor T Cells: Implications for Cancer Immunotherapy. Clin. Cancer Res. 2011, 17, 4975–4986.
- Maybruck, B.T.; Pfannenstiel, L.W.; Diaz-Montero, M.; Gastman, B.R. Tumor-derived exosomes induce CD8+ T cell suppressors. J. Immunother. Cancer 2017, 5, 65.
- Kim, J.W.; Wieckowski, E.; Taylor, D.D.; Reichert, T.E.; Watkins, S.; Whiteside, T.L. Fas ligand-positive membranous vesicles isolated from sera of patients with oral cancer induce apoptosis of activated T lymphocytes. Clin. Cancer Res. 2005, 11, 1010–1020.
- Abusamra, A.J.; Zhong, Z.; Zheng, X.; Li, M.; Ichim, T.E.; Chin, J.L.; Min, W.-P. Tumor exosomes expressing Fas ligand mediate CD8+ T-cell apoptosis. Blood Cells Mol. Dis. 2005, 35, 169–173.
- Salama, P.; Phillips, M.; Grieu, F.; Morris, M.; Zeps, N.; Joseph, D.; Platell, C.; Iacopetta, B. Tumor-Infiltrating FOXP3+ T Regulatory Cells Show Strong Prognostic Significance in Colorectal Cancer. J. Clin. Oncol. 2009, 27, 186–192.
- Agarwal, A.; Fanelli, G.; Letizia, M.; Tung, S.L.; Boardman, D.; Lechler, R.; Lombardi, G.; Smyth, L.A. Regulatory T Cell-Derived Exosomes: Possible Therapeutic and Diagnostic Tools in Transplantation. Front. Immunol. 2014, 5, 555.
- Greening, D.W.; Gopal, S.K.; Xu, R.; Simpson, R.J.; Chen, W. Exosomes and their roles in immune regulation and cancer. Semin. Cell Dev. Biol. 2015, 40, 72–81.
- Seo, N.; Akiyoshi, K.; Shiku, H. Exosome-mediated regulation of tumor immunology. Cancer Sci. 2018, 109, 2998–3004.
- Schwarzenbach, H.; Gahan, P.B. Exosomes in Immune Regulation. Noncoding RNA 2021, 7, 4.
- Li, J.; Huang, S.; Zhou, Z.; Lin, W.; Chen, S.; Chen, M.; Ye, Y. Exosomes derived from rAAV/AFP-transfected dendritic cells elicit specific T cell-mediated immune responses against hepatocellular carcinoma. Cancer Manag. Res. 2018, 10, 4945–4957.
- Lu, Z.; Zuo, B.; Jing, R.; Gao, X.; Rao, Q.; Liu, Z.; Qi, H.; Guo, H.; Yin, H. Dendritic cell-derived exosomes elicit tumor regression in autochthonous hepatocellular carcinoma mouse models. J. Hepatol. 2017, 67, 739–748.