Acute lymphoblastic leukemia (ALL) is the most common cancer in children and one of the success stories in cancer therapeutics. Risk-directed therapy based on clinical, biologic and genetic features has played a significant role in this accomplishment. Despite the observed improvement in survival rates, leukemia remains one of the leading causes of cancer-related deaths. Implementation of next-generation genomic and transcriptomic sequencing tools has illustrated the genomic landscape of ALL. However, the underlying dynamic changes at protein level still remain a challenge. Proteomics is a cutting-edge technology aimed at deciphering the mechanisms, pathways, and the degree to which the proteome impacts leukemia subtypes. Advances in mass spectrometry enable high-throughput collection of global proteomic profiles, representing an opportunity to unveil new biological markers and druggable targets.
- acute lymphoblastic leukemia
- proteomics
- biomarkers
1. Introduction
2. Molecular Basis of Acute Lymphoblastic Leukemia
3. The Era of Proteomics
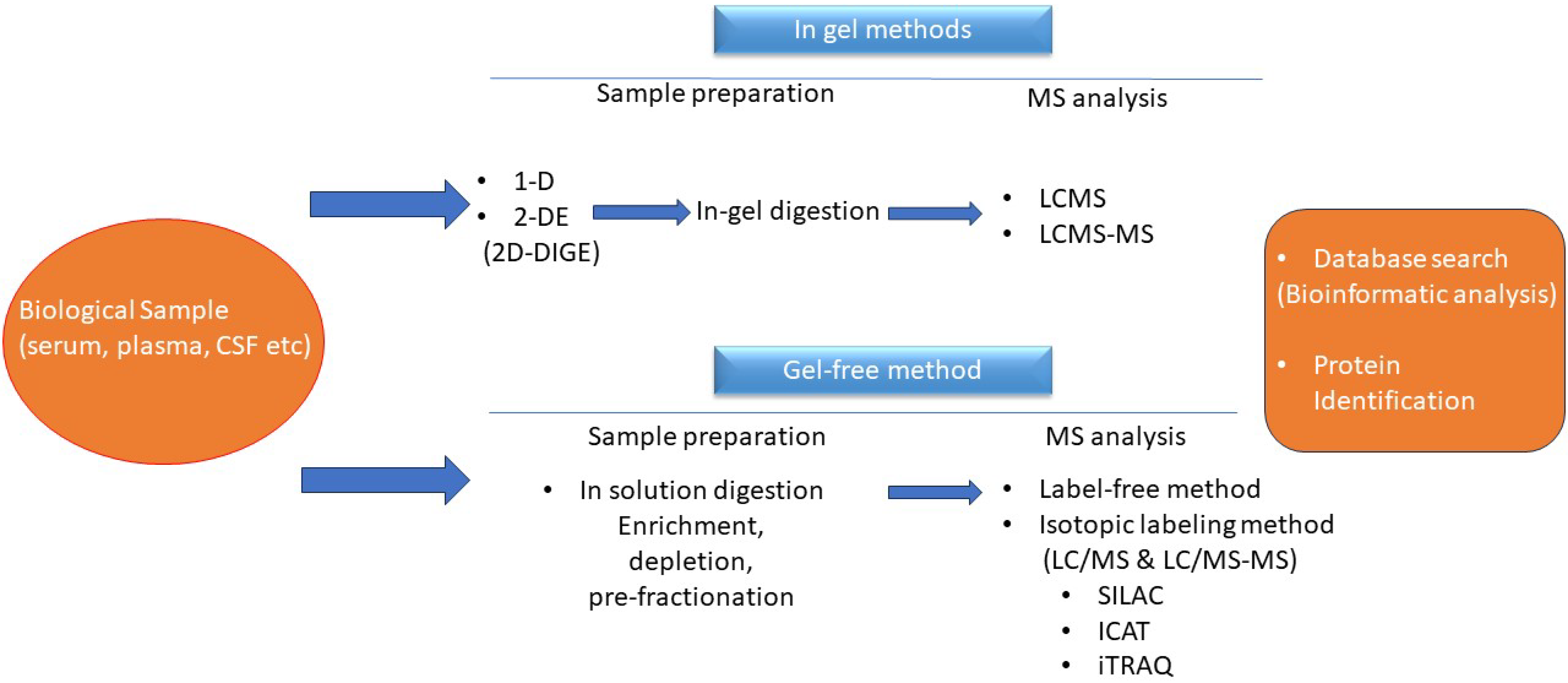
4. Application of Proteomics in Childhood ALL
5. Conclusions
This entry is adapted from the peer-reviewed paper 10.3390/diagnostics13172748
References
- Inaba, H.; Mullighan, C.G. Pediatric Acute Lymphoblastic Leukemia. Haematologica 2020, 105, 2524–2539.
- Iacobucci, I.; Mullighan, C.G. Genetic Basis of Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2017, 35, 975–983.
- Pui, C.-H.; Nichols, K.E.; Yang, J.J. Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat. Rev. Clin. Oncol. 2019, 16, 227–240.
- Jeha, S.; Pei, D.; Choi, J.; Cheng, C.; Sandlund, J.T.; Coustan-Smith, E.; Campana, D.; Inaba, H.; Rubnitz, J.E.; Ribeiro, R.C.; et al. Improved CNS Control of Childhood Acute Lymphoblastic Leukemia without Cranial Irradiation: St Jude Total Therapy Study 16. J. Clin. Oncol. 2019, 37, 3377–3391.
- Borowitz, M.J.; Wood, B.L.; Devidas, M.; Loh, M.L.; Raetz, E.A.; Salzer, W.L.; Nachman, J.B.; Carroll, A.J.; Heerema, N.A.; Gastier-Foster, J.M.; et al. Prognostic significance of minimal residual disease in high risk B-ALL: A report from Children’s Oncology Group study AALL0232. Blood 2015, 126, 964–971.
- Wood, B.; Wu, D.; Crossley, B.; Dai, Y.; Williamson, D.; Gawad, C.; Borowitz, M.J.; Devidas, M.; Maloney, K.W.; Larsen, E.; et al. Measurable residual disease detection by high-throughput sequencing improves risk stratification for pediatric B-ALL. Blood 2018, 131, 1350–1359.
- Smith, M.A.; Seibel, N.L.; Altekruse, S.F.; Ries, L.A.; Melbert, D.L.; O’Leary, M.; Smith, F.O.; Reaman, G.H. Outcomes for children and adolescents with cancer: Challenges for the twenty-first century. J. Clin. Oncol. 2010, 28, 2625–2634.
- Kwon, Y.W.; Jo, H.S.; Bae, S.; Seo, Y.; Song, P.; Song, M.; Yoon, J.H. Application of Proteomics in Cancer: Recent Trends and Approaches for Biomarkers Discovery. Front. Med. 2021, 8, 747333.
- Noone, A.M.; Cronin, K.A.; Altekruse, S.F.; Howlader, N.; Lewis, D.R.; Petkov, V.I.; Penberthy, L. Cancer Incidence and Survival Trends by Subtype Using Data from the Surveillance Epidemiology and End Results Program, 1992–2013. Cancer Epidemiol. Biomark. Prev. 2017, 26, 632–641.
- National Cancer Institute. Age-Adjusted and Age-Specific SEER Cancer Incidence Rates, 2014–2018. Available online: https://seer.cancer.gov/csr/1975_2018/results_merged/sect_02_childhood_cancer_iccc.pdf (accessed on 18 April 2021).
- Lim, J.Y.; Bhatia, S.; Robison, L.L.; Yang, J.J. Genomics of racial and ethnic disparities in childhood acute lymphoblastic leukemia. Cancer 2014, 120, 955–962.
- Wiemels, J. Perspectives on the causes of childhood leukemia. Chem. Biol. Interact. 2012, 196, 59–67.
- Stieglitz, E.; Loh, M.L. Genetic predispositions to childhood leukemia. Ther. Adv. Hematol. 2013, 4, 270–290.
- Buitenkamp, T.D.; Izraeli, S.; Zimmermann, M.; Forestier, E.; Heerema, N.A.; van den Heuvel-Eibrink, M.M.; Pieters, R.; Korbijn, C.M.; Silverman, L.B.; Schmiegelow, K.; et al. Acute lymphoblastic leukemia in children with Down syndrome: A retrospective analysis from the Ponte di Legno study group. Blood 2014, 123, 70–77.
- Mullighan, C.G.; Collins-Underwood, J.R.; Phillips, L.A.; Loudin, M.G.; Liu, W.; Zhang, J.; Ma, J.; Coustan-Smith, E.; Harvey, R.C.; Willman, C.L.; et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat. Genet. 2009, 41, 1243–1246.
- Lilljebjörn, H.; Fioretos, T. New oncogenic subtypes in pediatric B-cell precursor acute lymphoblastic leukemia. Blood 2017, 130, 1395–1401.
- Bastian, L.; Schroeder, M.P.; Eckert, C.; Schlee, C.; Tanchez, J.O.; Kämpf, S.; Wagner, D.L.; Schulze, V.; Isaakidis, K.; Lázaro-Navarro, J.; et al. PAX5 biallelic genomic alterations define a novel subgroup of B-cell precursor acute lymphoblastic leukemia. Leukemia 2019, 33, 1895–1909.
- Gu, Z.; Churchman, M.; Roberts, K.; Li, Y.; Liu, Y.; Harvey, R.C.; McCastlain, K.; Reshmi, S.C.; Payne-Turner, D.; Iacobucci, I.; et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat. Commun. 2016, 7, 13331.
- Paulsson, K.; Lilljebjörn, H.; Biloglav, A.; Olsson, L.; Rissler, M.; Castor, A.; Barbany, G.; Fogelstrand, L.; Nordgren, A.; Sjögren, H.; et al. The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nat. Genet. 2015, 47, 672–676.
- Vora, A.; Goulden, N.; Wade, R.; Mitchell, C.; Hancock, J.; Hough, R.; Rowntree, C.; Richards, S. Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): A randomised controlled trial. Lancet Oncol. 2013, 14, 199–209.
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552.
- Pui, C.H.; Yang, J.J.; Hunger, S.P.; Pieters, R.; Schrappe, M.; Biondi, A.; Vora, A.; Baruchel, A.; Silverman, L.B.; Schmiegelow, K.; et al. Childhood Acute Lymphoblastic Leukemia: Progress Through Collaboration. J. Clin. Oncol. 2015, 3, 2938–2948.
- Harrison, C.J. Cytogenetics of paediatric and adolescent acute lymphoblastic leukaemia. Br. J. Haematol. 2009, 144, 147–156.
- Greaves, M. Darwin and evolutionary tales in leukemia. The Ham-Wasserman Lecture. Hematol. Am. Soc. Hematol. Educ. Program. 2009, 2009, 3–12.
- Lilljebjörn, H.; Henningsson, R.; Hyrenius-Wittsten, A.; Olsson, L.; Orsmark-Pietras, C.; von Palffy, S.; Askmyr, M.; Rissler, M.; Schrappe, M.; Cario, G.; et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat. Commun. 2016, 7, 11790.
- Felice, M.S.; Gallego, M.S.; Alonso, C.N.; Alfaro, E.M.; Guitter, M.R.; Bernasconi, A.R.; Rubio, P.L.; Zubizarreta, P.A.; Rossi, J.G. Prognostic impact of t(1;19)/ TCF3-PBX1 in childhood acute lymphoblastic leukemia in the context of Berlin-Frankfurt-Münster-based protocols. Leuk. Lymphoma 2011, 52, 1215–1221.
- Jeha, S.; Pei, D.; Raimondi, S.C.; Onciu, M.; Campana, D.; Cheng, C.; Sandlund, J.T.; Ribeiro, R.C.; Rubnitz, J.E.; Howard, S.C.; et al. Increased risk for CNS relapse in pre-B cell leukemia with the t(1;19)/TCF3-PBX1. Leukemia 2009, 23, 1406–1409.
- Mullighan, C.G. Molecular genetics of B-precursor acute lymphoblastic leukemia. J. Clin. Investig. 2012, 122, 3407–3415.
- Kang, Z.J.; Liu, Y.F.; Xu, L.Z.; Long, Z.J.; Huang, D.; Yang, Y.; Liu, B.; Feng, J.X.; Pan, Y.J.; Yan, J.S.; et al. The Philadelphia chromosome in leukemogenesis. Chin. J. Cancer 2016, 35, 48.
- Biondi, A.; Gandemer, V.; De Lorenzo, P.; Cario, G.; Campbell, M.; Castor, A.; Pieters, R.; Baruchel, A.; Vora, A.; Leoni, V.; et al. Imatinib treatment of paediatric Philadelphia chromosome-positive acute lymphoblastic leukaemia (EsPhALL2010): A prospective, intergroup, open-label, single-arm clinical trial. Lancet Haematol. 2018, 5, e641–e652.
- Schultz, K.R.; Carroll, A.; Heerema, N.A.; Bowman, W.P.; Aledo, A.; Slayton, W.B.; Sather, H.; Devidas, M.; Zheng, H.W.; Davies, S.M.; et al. Children’s Oncology Group. Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children’s Oncology Group study AALL0031. Leukemia 2014, 28, 1467–1471.
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N. Engl. J. Med. 2014, 371, 1005–1015.
- Ding, Y.Y.; Stern, J.W.; Jubelirer, T.F.; Wertheim, G.B.; Lin, F.; Chang, F.; Gu, Z.; Mullighan, C.G.; Li, Y.; Harvey, R.C.; et al. Clinical efficacy of ruxolitinib and chemotherapy in a child with Philadelphia chromosome-like acute lymphoblastic leukemia with GOLGA5-JAK2 fusion and induction failure. Haematologica 2018, 103, e427–e431.
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211–1218.
- D’Angiò, M.; Valsecchi, M.G.; Testi, A.M.; Conter, V.; Nunes, V.; Parasole, R.; Colombini, A.; Santoro, N.; Varotto, S.; Caniglia, M.; et al. Clinical features and outcome of SIL/TAL1-positive T-cell acute lymphoblastic leukemia in children and adolescents: A 10-year experience of the AIEOP group. Haematologica 2015, 100, e10–e13.
- Girardi, T.; Vicente, C.; Cools, J.; De Keersmaecker, K. The genetics and molecular biology of T-ALL. Blood 2017, 129, 1113–1123.
- Patrick, K.; Vora, A. Update on biology and treatment of T-cell acute lymphoblastic leukaemia. Curr. Opin. Pediatr. 2015, 27, 44–49.
- Waanders, E.; Gu, Z.; Dobson, S.M.; Antić, Ž.; Crawford, J.C.; Ma, X.; Edmonson, M.N.; Payne-Turner, D.; van de Vorst, M.; Jongmans, M.C.J.; et al. Mutational landscape and patterns of clonal evolution in relapsed pediatric acute lymphoblastic leukemia. Blood Cancer Discov. 2020, 1, 96–111.
- Burke, M.J.; Kostadinov, R.; Sposto, R.; Gore, L.; Kelley, S.M.; Rabik, C.; Trepel, J.B.; Lee, M.J.; Yuno, A.; Lee, S.; et al. Decitabine and Vorinostat with Chemotherapy in Relapsed Pediatric Acute Lymphoblastic Leukemia: A TACL Pilot Study. Clin. Cancer Res. 2020, 26, 2297–2307.
- Place, A.E.; Pikman, Y.; Stevenson, K.E.; Harris, M.H.; Pauly, M.; Sulis, M.L.; Hijiya, N.; Gore, L.; Cooper, T.M.; Loh, M.L.; et al. Phase I trial of the mTOR inhibitor everolimus in combination with multi-agent chemotherapy in relapsed childhood acute lymphoblastic leukemia. Pediatr. Blood Cancer 2018, 65, e27062.
- Altelaar, A.F.; Munoz, J.; Heck, A.J. Next-generation proteomics: Towards an integrative view of proteome dynamics. Nat. Rev. Genet. 2013, 14, 35–48.
- Macklin, A.; Khan, S.; Kislinger, T. Recent advances in mass spectrometry based clinical proteomics: Applications to cancer research. Clin. Proteom. 2020, 24, 17.
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550.
- Aebersold, R.; Agar, J.N.; Amster, I.J.; Baker, M.S.; Bertozzi, C.R.; Boja, E.S.; Costello, C.E.; Cravatt, B.F.; Fenselau, C.; Garcia, B.A.; et al. How many human proteoforms are there? Nat. Chem. Biol. 2018, 14, 206–214.
- Melani, R.D.; Gerbasi, V.R.; Anderson, L.C.; Sikora, J.W.; Toby, T.K.; Hutton, J.E.; Butcher, D.S.; Negrão, F.; Seckler, H.S.; Srzentić, K.; et al. The Blood Proteoform Atlas: A reference map of proteoforms in human hematopoietic cells. Science 2022, 28, 411–418.
- Cunningham, R.; Ma, D.; Li, L. Mass Spectrometry-based Proteomics and Peptidomics for Systems Biology and Biomarker Discovery. Front. Biol. 2012, 1, 313–335.
- Pursiheimo, A.; Vehmas, A.P.; Afzal, S.; Suomi, T.; Chand, T.; Strauss, L.; Poutanen, M.; Rokka, A.; Corthals, G.L.; Elo, L.L. Optimization of Statistical Methods Impact on Quantitative Proteomics Data. J. Proteome Res. 2015, 14, 4118–4126.
- Pietrowska, M.; Wlosowicz, A.; Gawin, M.; Widlak, P. MS-Based Proteomic Analysis of Serum and Plasma: Problem of High Abundant Components and Lights and Shadows of Albumin Removal. Adv. Exp. Med. Biol. 2019, 1073, 57–76.
- Dunphy, K.; O’Mahoney, K.; Dowling, P.; O’Gorman, P.; Bazou, D. Clinical Proteomics of Biofluids in Haematological Malignancies. Int. J. Mol. Sci. 2021, 22, 8021.
- Xiao, Z.; Conrads, T.P.; Lucas, D.A.; Janini, G.M.; Schaefer, C.F.; Buetow, K.H.; Issaq, H.J.; Veenstra, T.D. Direct ampholyte-free liquid-phase isoelectric peptide focusing: Application to the human serum proteome. Electrophoresis 2004, 25, 128–133.
- Zecha, J.; Gabriel, W.; Spallek, R.; Chang, Y.C.; Mergner, J.; Wilhelm, M.; Bassermann, F.; Kuster, B. Linking post-translational modifications and protein turnover by site-resolved protein turnover profiling. Nat. Commun. 2022, 13, 165.
- Bantscheff, M.; Schirle, M.; Sweetman, G.; Rick, J.; Kuster, B. Quantitative mass spectrometry in proteomics: A critical review. Anal. Bioanal. Chem. 2007, 389, 1017–1031.
- Zhu, W.; Smith, J.W.; Huang, C.M. Mass spectrometry-based label-free quantitative proteomics. J. Biomed. Biotechnol. 2010, 2010, 840518.
- Calderon-Rodríguez, S.I.; Sanabria-Salas, M.C.; Umaña-Perez, A. A comparative proteomic study of plasma in Colombian childhood acute lymphoblastic leukemia. PLoS ONE 2019, 14, e0221509.
- Wang, D.; Lv, Y.Q.; Liu, Y.F.; Du, X.J.; Li, B. Differential protein analysis of lymphocytes between children with acute lymphoblastic leukemia and healthy children. Leuk. Lymphoma 2013, 54, 381–386.
- Cavalcante Mde, S.; Torres-Romero, J.C.; Lobo, M.D.; Moreno, F.B.; Bezerra, L.P.; Lima, D.S.; Matos, J.C.; Moreira Rde, A.; Monteiro-Moreira, A.C. A panel of glycoproteins as candidate biomarkers for early diagnosis and treatment evaluation of B-cell acute lymphoblastic leukemia. Biomark. Res. 2016, 27, 4.
- Yu, R.; Yang, S.; Liu, Y.; Zhu, Z. Identification and validation of serum autoantibodies in children with B-cell acute lymphoblastic leukemia by serological proteome analysis. Proteome Sci. 2022, 20, 3.
- Guzmán-Ortiz, A.L.; Aparicio-Ozores, G.; Valle-Rios, R.; Medina-Contreras, O.; Patiño-López, G.; Quezada, H. Proteomic changes in a childhood acute lymphoblastic leukemia cell line during the adaptation to vincristine. Bol. Med. Hosp. Infant. Mex. 2017, 74, 181–192.
- Risinger, A.L.; Giles, F.J.; Mooberry, S.L. Microtubule dynamics as a target in oncology. Cancer Treat. Rev. 2009, 35, 255–261.
- Kemper, E.K.; Zhang, Y.; Dix, M.M.; Cravatt, B.F. Global profiling of phosphorylation-dependent changes in cysteine reactivity. Nat. Methods 2022, 19, 341–352.
- Floyd, B.M.; Drew, K.; Marcotte, E.M. Systematic Identification of Protein Phosphorylation-Mediated Interactions. J. Proteome Res. 2021, 20, 1359–1370.
- de Smith, A.J.; Ojha, J.; Francis, S.S.; Sanders, E.; Endicott, A.A.; Hansen, H.M.; Smirnov, I.; Termuhlen, A.M.; Walsh, K.M.; Metayer, C.; et al. Clonal and microclonal mutational heterogeneity in high hyperdiploid acute lymphoblastic leukemia. Oncotarget 2016, 7, 72733–72745.
- Malinowska-Ozdowy, K.; Frech, C.; Schönegger, A.; Eckert, C.; Cazzaniga, G.; Stanulla, M.; zur Stadt, U.; Mecklenbräuker, A.; Schuster, M.; Kneidinger, D.; et al. KRAS and CREBBP mutations: A relapse-linked malicious liaison in childhood high hyperdiploid acute lymphoblastic leukemia. Leukemia 2015, 29, 1656–1667.
- Vainchenker, W.; Constantinescu, S.N. JAK/STAT signaling in hematological malignancies. Oncogene 2013, 32, 2601–2613.
- Liu, X.; Qu, C.K. Protein Tyrosine Phosphatase SHP-2 (PTPN11) in Hematopoiesis and Leukemogenesis. J. Signal Transduct. 2011, 2011, 195239.
- Zhou, B.; Lin, W.; Long, Y.; Yang, Y.; Zhang, H.; Wu, K.; Chu, Q. Notch signaling pathway: Architecture, disease, and therapeutics. Signal Transduct Target Ther. 2022, 7, 95.
- Yuan, X.; Wu, H.; Xu, H.; Xiong, H.; Chu, Q.; Yu, S.; Wu, G.S.; Wu, K. Notch signaling: An emerging therapeutic target for cancer treatment. Cancer Lett. 2015, 369, 20–27.
- Cordo’, V.; Meijer, M.T.; Hagelaar, R.; de Goeij-de Haas, R.R.; Poort, V.M.; Henneman, A.A.; Piersma, S.R.; Pham, T.V.; Oshima, K.; Ferrando, A.A.; et al. Phosphoproteomic profiling of T cell acute lymphoblastic leukemia reveals targetable kinases and combination treatment strategies. Nat. Commun. 2022, 13, 1048.
- Beekhof, R.; van Alphen, C.; Henneman, A.A.; Knol, J.C.; Pham, T.V.; Rolfs, F.; Labots, M.; Henneberry, E.; Le Large, T.Y.; de Haas, R.R.; et al. INKA, an integrative data analysis pipeline for phosphoproteomic inference of active kinases. Mol. Syst. Biol. 2019, 15, e8250.
- Leo, I.R.; Aswad, L.; Stahl, M.; Kunold, E.; Post, F.; Erkers, T.; Struyf, N.; Mermelekas, G.; Joshi, R.N.; Gracia-Villacampa, E.; et al. Integrative multi-omics and drug response profiling of childhood acute lymphoblastic leukemia cell lines. Nat. Commun. 2022, 13, 1691.
- Baytan, B.; Evim, M.S.; Güler, S.; Güneş, A.M.; Okan, M. Acute Central Nervous System Complications in Pediatric Acute Lymphoblastic Leukemia. Pediatr. Neurol. 2015, 53, 312–318.
- Manley, S.; Keenan, R.; Campbell, H.; Caswell, M.; Pizer, B. No evidence for routine cerebrospinal fluid cytology in detecting asymptomatic central nervous system relapse in children with acute lymphoblastic leukaemia: 20 years’ experience of a UK primary treatment centre. Br. J. Haematol. 2014, 164, 462–464.
- Thastrup, M.; Duguid, A.; Mirian, C.; Schmiegelow, K.; Halsey, C. Central nervous system involvement in childhood acute lymphoblastic leukemia: Challenges and solutions. Leukemia 2022, 36, 2751–2768.
- Galicia, N.; Díez, P.; Dégano, R.M.; Guest, P.C.; Ibarrola, N.; Fuentes, M. Proteomic Biomarker Identification in Cerebrospinal Fluid for Leptomeningeal Metastases with Neurological Complications. Adv. Exp. Med. Biol. 2017, 974, 85–96.
- Roy, S.; Josephson, S.A.; Fridlyand, J.; Karch, J.; Kadoch, C.; Karrim, J.; Damon, L.; Treseler, P.; Kunwar, S.; Shuman, M.A.; et al. Protein biomarker identification in the CSF of patients with CNS lymphoma. J. Clin. Oncol. 2008, 26, 96–105.
- Carbonara, K.; Andonovski, M.; Coorssen, J.R. Proteomes Are of Proteoforms: Embracing the Complexity. Proteomes 2021, 9, 38.
- Priola, G.M.; Foster, M.W.; Deal, A.M.; Richardson, B.M.; Thompson, J.W.; Blatt, J. Cerebrospinal fluid proteomics in children during induction for acute lymphoblastic leukemia: A pilot study. Pediatr. Blood Cancer 2015, 62, 1190–1194.