Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Liver regeneration has been studied for many decades, and the mechanisms underlying regeneration of normal liver following resection are well described. However, no less relevant is the study of mechanisms that disrupt the process of liver regeneration. First of all, a violation of liver regeneration can occur in the presence of concomitant hepatic pathology, which is a key factor reducing the liver’s regenerative potential. Understanding these mechanisms could enable the rational targeting of specific therapies to either reduce the factors inhibiting regeneration or to directly stimulate liver regeneration.
- liver regeneration
- liver pathology
- metabolic mechanisms
- FLIM
1. Introduction
The most effective treatment for neoplasms localized in the liver (e.g., tumor, metastases) is resection and relies on the high regenerative potential of this organ. It is known that 25% is the minimum amount of hepatic remnant sufficient for adequate recovery in a patient with a morphologically unchanged liver. In the presence of drug-induced damage or hepatic pathology, there is a requirement for the liver remnant to be at least 40% of the initial volume [1]. When the volume of the resected part of the organ exceeds this limit, the liver is not able to restore sufficient function. However, even in cases of the removal of smaller fragments of the liver, there is a risk of fatal, acute post-resection liver failure, which occurs in 5–8% of patients and remains the main cause of mortality in liver surgery [1][2][3][4][5][6].
The following sections consider the main hepatic pathologies that are widespread in patients and describe the principal metabolic mechanisms that impair both liver function and its regenerative capacity.
2. Non-Alcoholic Fatty Liver Disease
There has been an explosion of interest in non-alcoholic fatty liver disease (NAFLD) and its more advanced stage, NASH, because of their growing impact on world health. Globally, the prevalence of NAFLD is estimated at 25% of the population, while 25% of NAFLD patients are estimated to have NASH. NAFLD is a general term that includes conditions that vary in the severity of injury and degree of fibrosis. The presence of these pathological conditions dramatically increases the risk of developing cirrhosis and has caused an alarming increase in hepatocellular carcinoma [7][8]. The mechanisms of the pathogenesis of NAFLD and NASH are shown in the Figure 1.
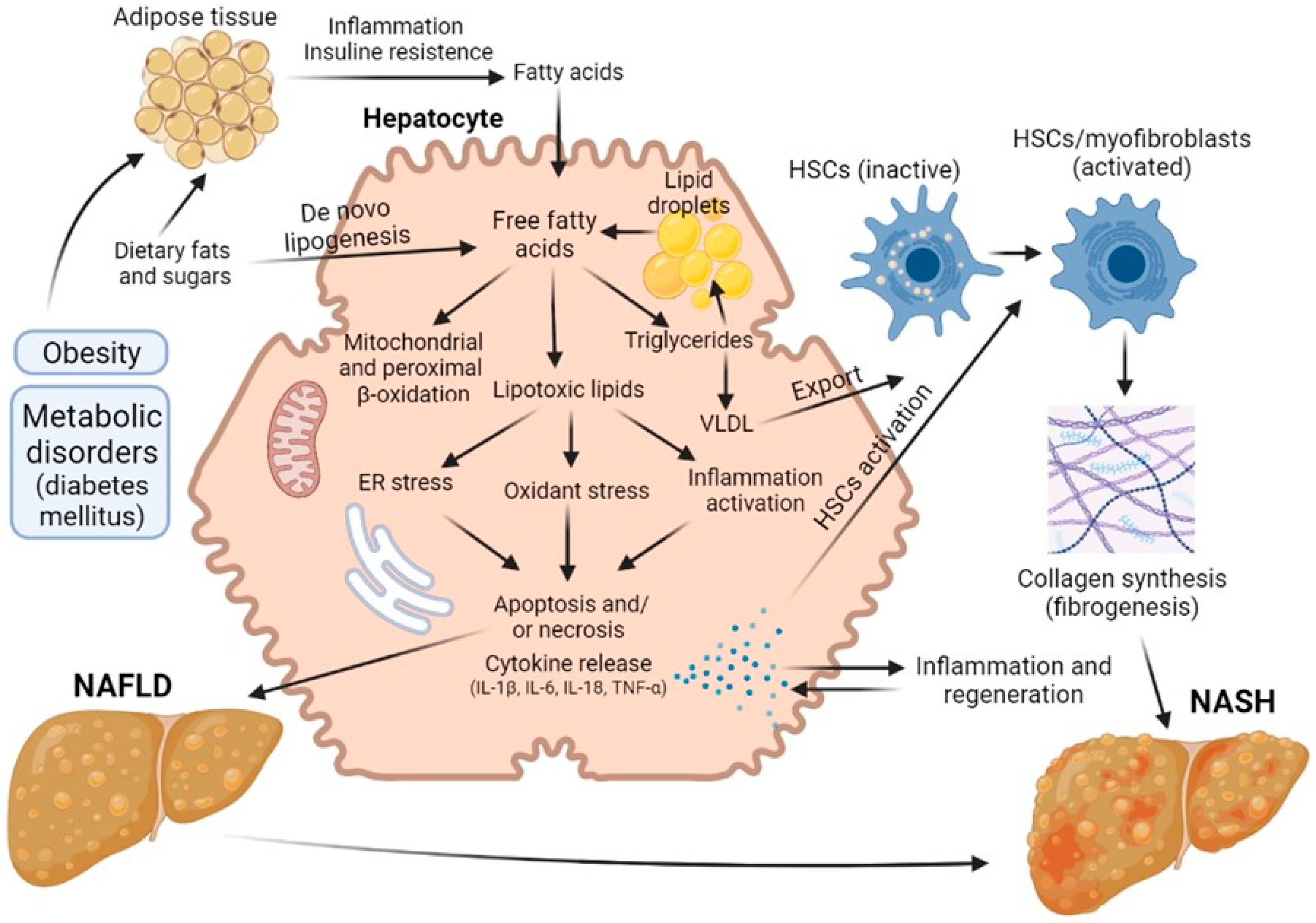
Figure 1. Road map of the pathological mechanisms of NAFLD and NASH.
Hepatic steatosis is the most common form of non-alcoholic fatty liver disease and is diagnosed when more than 5% of hepatocytes contain lipid droplets rich in triacylglycerol (TAG) in the absence of a secondary factor such as excessive alcohol intake, viral infection or drug damage [9][10]. Free fatty acids (FFAs) can be obtained from diet, adipose tissue lipolysis and/or de novo lipogenesis. They are then oxidized via β-oxidation, esterified to TAGs and packaged into lipoproteins, which are either secreted or stored as lipid droplets. NASH, as opposed to steatosis, is typically accompanied by pericellular fibrosis, which may progress to cirrhosis [11].
Initially, it was believed that the synthesis of triacylglycerol and the accumulation of fat in the liver was a hepatoprotective mechanism, but later data appeared indicating that such excess content of lipids in the liver cells is a risk factor for the progression of liver disease [9][10]. Steatosis was not diagnosed until 1980, but it is now believed that the presence of steatosis is a risk factor that worsens the prognosis of liver regeneration after resection. Today, about 20% of liver resection patients and up to 25% of liver transplant donors have some degree of steatosis. In a number of studies, it has been demonstrated that the risk of postoperative complications and lethal outcomes grows with increasing severity of steatosis [9][10][12][13][14][15]. In addition, patients with hepatic steatosis are particularly vulnerable to ischemia/reperfusion injury, which can often occur during surgery and liver transplantation [16].
These days, NAFLD, and in particular steatosis, is recognized as a risk factor for resection, although the exact mechanisms of this issue has not yet been established. A number of authors have proposed a sequence known as the “two-hit theory”, which suggests a two-stage process of disease development [17][18]. As stated above, at the first stage, the initial pathological changes are caused by an excess of FFAs in the liver, which are then metabolized to TAG [13][19]. Such processes make the liver vulnerable to aggressive factors at the second stage, caused by oxidative stress and the action of pro-inflammatory cytokines [20]. The ‘two-hit’ theory was posited for several years. However, this view is now considered outdated. There are many molecular pathways that contribute to the development of NAFLD and NASH, and it is not even certain whether NASH is always preceded by NAFLD. In defining the pathogenic drivers of NAFLD and NASH, a useful conceptual framework is that the liver’s capacity to handle the primary metabolic energy substrates, carbohydrates and fatty acids is overwhelmed, leading to the accumulation of toxic lipid species [21][22][23]. These metabolites induce hepatocellular stress, injury and death, leading to fibrogenesis and genomic instability that predispose to cirrhosis and hepatocellular carcinoma. Thus, when fatty acids are either supplied in excess, or their disposal is impaired, they may serve as substrates for the generation of lipotoxic species that provoke oxidative stress and hepatocellular injury.
This chain of events leads to the development of a key factor in the pathogenesis of steatosis—mitochondrial dysfunction [24][25], an event causing a decrease in the regenerative potential of the liver. In a study comparing patients with low (3%) and high (>17%—moderate steatosis) levels of intrahepatic triacylglycerol, it was found that in the presence of steatosis, the rates of lipolysis and of gluconeogenesis increased by 50% and by 30%, respectively. Normally, long-chain fatty acids are metabolized into acyl-CoA molecules by specific acyl-CoA synthases and then enter the mitochondrial matrix. At the same time, reduced NADH, formed as a result of β-oxidation and the operation of the tricarboxylic acid cycle, deliver their electrons and protons to the respiratory chain of mitochondria with the formation of NAD+ and adenosine triphosphate (ATP) [26]. Excessive influx of fatty acids into the liver leads to an overload of the mitochondria, resulting in an accumulation of incompletely oxidized substrates, with increased formation of ROS [27][28]. Chronic oxidative stress leads to the depletion of antioxidants such as glutathione, thereby further compounding cell damage [7]. In addition, persistent oxidative stress leads to disruption of the integrity of the mitochondrial membrane and, therefore, to mitochondrial dysfunction, which is defined by a number of authors as a key event triggering an irreversible cascade of damage to liver cells. As a result, the production of mitochondrial ATP in fatty hepatocytes decreases, and the overall energy metabolism in the cells is disrupted [13][29][30], this representing the main contributory factor in the decrease in regenerative capacity of the liver in the presence of this pathology.
3. Diabetes-Provided Fatty Liver Disease
Aside from being caused by excessive consumption of dietary fat, fatty liver develops with metabolic disorders—in particular, diabetes mellitus of types I and II. Diabetes is a chronic metabolic disorder with a rapidly increasing prevalence. It alters the carbohydrate, lipid and protein metabolisms of patients. The World Health Organization predicts that 300 million people around the world will suffer from diabetes mellitus by 2025 [31].
Diabetes mellitus has the clearest biological link to the progression of NAFLD, and up to 75% of individuals with diabetes have NAFLD; subsequently, 2–3% of patients develop NASH [7][32][33][34]. The development of fatty liver disease, combined with concomitant hypertension, a characteristic of diabetes mellitus, sharply reduces the regenerative potential of the liver. Diabetes mellitus is also considered a risk factor for prognosis after liver resection in patients with hepatocellular carcinoma, and postoperative morbidity is more common among diabetic patients than among nondiabetic patients. Several critical pathways have been identified as causing liver damage in diabetic patients. Insulin resistance, the main cause of hyperglycaemia and compensatory hyperinsulinaemia, is the predominant cause of impaired regenerative response [33][35]. It has been shown that streptozotocin-induced diabetic rats subjected to PH present with a decrease in the level of proliferating cell nuclear antigen and a significant decrease in cyclin D1 levels, suggesting that few hepatocytes are capable of entering the cell cycle [36]. The reasons are that diabetes is generally followed by increased free radical production or reduced antioxidant protection, leading to lipid peroxidation. Such streptozotocin-induced diabetic rats were also found to present with an increase in •OH production, which could result in DNA damage. Hyperglycemia in these rats leads to an increase in hepatic ROS production and is further exacerbated after PH. Oxygen free radicals are extensively formed in diabetic patients by glucose oxidation, non-enzymatic protein glycation and subsequent oxidative degradation of the glycated proteins [33]. Hyperglycemia-induced oxidative stress, followed by derangement of protein, carbohydrate and lipid metabolism, thereby leads to increased oxidative stress and to further triggering of the inflammatory cascade. Both oxidative stress and inflammatory responses act as damaging agents in aggravating the pathological condition of diabetes [37]. Eventually, such violation of the metabolism of liver cells will inevitably disrupt the regenerative capacity of the liver.
4. Fibrosis and Cirrhosis
Liver fibrosis, or its decompensated form, cirrhosis, develops in response to chronic liver damage of various origins. Fibrosis is characterized by excessive formation and deposition of extracellular matrix and collagen [7], accompanied by a violation of tissue architecture, the development of portal hypertension and cellular hypoxia. The progression of fibrosis results from chronic damage and chronic recovery process in the liver. In cases of acute, rather than severe, liver damage, the remaining mature hepatocytes are able to replace apoptotic and necrotic cells [38], but in chronic damage, the regenerative process is disrupted, and hepatocytes are replaced by extracellular matrix proteins [39][40]. A key event in liver fibrosis is the activation of HSCs, resulting in these cells acquiring a myofibroblast-like phenotype characterized by active proliferation, loss of vitamin A stores and activation of the production and release of alpha-smooth muscle actin and collagen (types I and III) into the extracellular space [41]. In addition, upregulation of a tissue inhibitor of metalloproteinase-1 in the fibrotic liver contributes to collagen deposition by inhibiting the dissolution of the extracellular matrix. Persistent production of growth factors for HSCs, fibrogenic cytokines and chemokines by various types of liver cells is involved in fibrogenesis in chronic inflammation. Among these, TGF-ß, produced by immune cells, directly promotes fibrogenesis by inducing the transcription of types I and III collagen through the Smad signaling pathway [42]. IL-1ß and TNF-α do not induce HSC activation but do contribute to liver fibrosis by, instead, mediating the survival of the activated HSCs [42].
As mentioned earlier, the polyploidy of liver cells plays an important role in ensuring the efficiency of regeneration. However, in chronic diseases that are characterized by compensatory regeneration (in particular, liver cirrhosis), the number of polyploid cells increases by 20%. The frequency of polyploid mononuclear cells and binuclear hepatocytes increases in direct proportion to the severity of fibrosis [43][44]. When chronic loss of hepatocytes and activation of HSCs leads to an abnormal tissue environment, the chronic compensatory regeneration of hepatocytes often has negative consequences, including the development of neoplasia [45]. The mechanisms of the pathogenesis of liver fibrosis are shown in Figure 2.
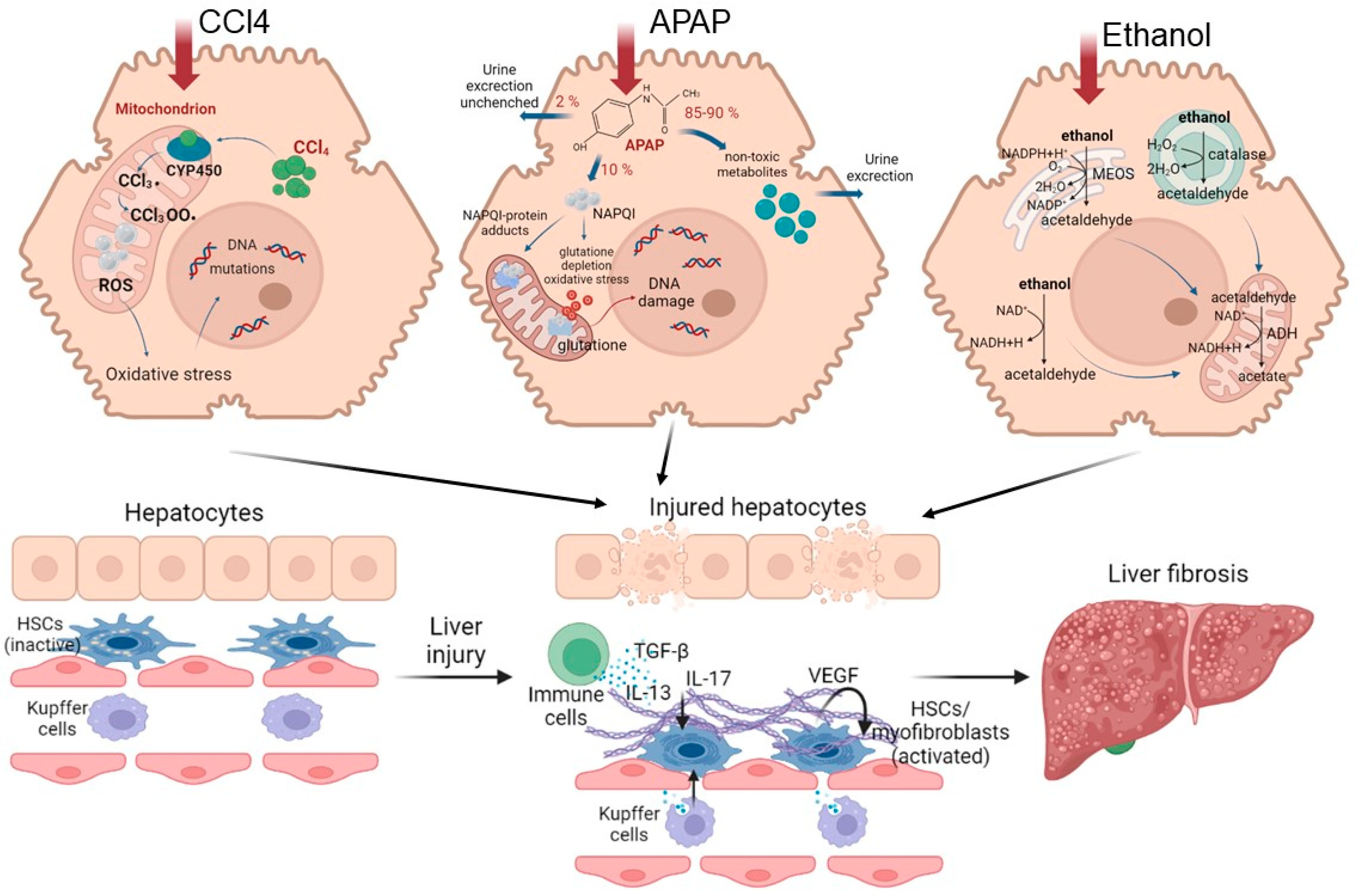
Figure 2. Road map of pathological mechanisms of liver fibrosis when exposed to various damaging agents.
Understanding the mechanisms of the pathogenesis of fibrosis has been achieved using various models. CCl4 is widely used to model chronic liver fibrosis and cirrhosis. In hepatocytes, the toxin is metabolized with the participation of cytochrome P450, resulting in the formation of the highly reactive free radicals CCl3• and CCl3O2•. These free radicals activate the fatty acid peroxidation reactions of the mitochondrial membrane and disrupt the integrity and stability of the mitochondrial structure, leading to mitochondrial dysfunction and, ultimately, to necrosis of hepatocytes in the central and parts of the intermediate zones of all liver lobules [46][47][48]. Normally, in the mitochondrial respiration reactions used for energy production, a significant number of electrons are transferred to ATP, while only a few electrons reduced with the formation of ROS. When exposed to CCl4, the structure and functions of the mitochondria are disrupted [49], causing additional generation of ROS. As a result, such a cascade of events leads to the death of hepatocytes by necrosis, thereby affecting liver function. With regular repeated exposure to the toxin, a constant inflammatory and pathological regenerative process is maintained, characterized by the development of chronic regeneration nodes, which ultimately leads to the active accumulation of collagen fibers in response to such chronic damage.
Another commonly used model for the induction of fibrosis and cirrhosis is acetaminophen (APAP) administration. Toxicity due to APAP overdose may arise as a consequence either of an acute overdose or from repeated/staggered dosing over a short period of time. Briefly, APAP hepatotoxicity can be divided into three phases. During the initiation phase after an overdose, APAP is rapidly metabolized to its reactive metabolite, N-acetyl-p-benzoquinone imine (NAPQI), which is removed by glutathione conjugation, leading to rapid depletion of the cellular glutathione stores. The excess NAPQI forms cellular protein adducts, particularly in mitochondria, leading to mitochondrial dysfunction and to the generation of ROS [50]. A previous study utilizing incremental doses of APAP in mice showed that liver regeneration after APAP toxicity was dose dependent. Low overdose of APAP in mice (300 mg/kg) caused not only extensive liver injury but also significant compensatory regeneration, leading to regression of the injury and spontaneous recovery. However, after a severe overdose of APAP (600 mg/kg), liver regeneration was remarkably inhibited, resulting in sustained injury and decreased survival. It is interesting that the marked inhibition of regeneration at the higher dose was not due to a lack of critical liver mass, as >50% of hepatocytes were still viable at this dose, even at peak injury. In fact, peak injury was not remarkably different between the two doses, whereas regeneration was significantly impaired only at the higher dose [51]. The injury phases of APAP hepatotoxicity are subsequently followed by a recovery phase, in which compensatory hepatocellular proliferation is initiated; dead cells are replaced by newly formed cells, leading to liver regeneration and recovery. In cases in which a robust liver-regeneration response is initiated, liver injury is resolved, and liver function is restored spontaneously. In cases in which liver regeneration fails, acute liver injury can progress to acute liver failure, with multi-organ failure and death. Comprehensive analysis of the signaling pathways revealed that several pro- and anti-regenerative pathways were differentially affected in a dose-dependent manner. APAP-stimulated liver injury is characterized by considerably increased expression of IL-1β, IL-18 and the levels of other inflammatory dependent mediators. Paradoxically, dose-dependent activation of EGFR was observed after APAP overdose, such that the activation of EGFR was greater at the higher doses of APAP, where liver regeneration was inhibited. Whereas early inhibition of EGFR activation by pharmacological intervention in mice remarkably attenuated APAP hepatotoxicity, delayed inhibition of EGFR activation led to impaired compensatory liver regeneration, suggesting a dual role of EGFR in both injury initiation and the subsequent liver regeneration after APAP overdose [52]. Elevated plasma levels of inactive, single-chain HGF have also been observed in patients who overdose on APAP. Levels of the angiogenic factor VEGF and expression of its receptors VEGFR1, VEGFR2 and VEGFR3 also increase in mouse liver after paracetamol overdose. The expression of cytokines (TNF and IL-6) also increases after APAP overdose in mice. The foregoing indicates a compensatory activation of pathological recovery processes.
5. Alcoholic Liver Disease
Another common pathological condition is alcoholic fatty liver disease (ALD), caused by chronic alcohol (ethanol) abuse. A subset of patients with ALD will progress to develop alcohol steatohepatitis and then fibrosis if they continue to consume alcohol heavily [53]. In the later stages, ALD is characterized by a marked fibrotic response and the development of advanced fibrosis, which is associated with early mortality. The pattern of fibrosis in ALD is characterized by pericellular and perisinusoidal (terminal small blood vessels with fenestrated discontinuous epithelium in the liver) matrix accumulation. When fibrosis becomes advanced, the liver becomes cirrhotic and consists predominantly of fibrotic tissue, which leads to a major disturbance of hepatic blood flow due to narrowing of the vascular structures within the hepatic lobules, including the sinusoids. In addition, the function of the liver decreases owing to the loss of hepatocytes [53][54].
This scenario of the development of the disease is due to the specifics of ethanol metabolism in hepatocytes. The oxidative pathways of alcohol metabolism involve three enzymes: alcohol dehydrogenase (ADH) in the cytosol, which converts alcohol to acetaldehyde; at elevated ethanol concentrations, CYP2E1 in the microsomes, which also assumes an important role in metabolizing ethanol to acetaldehyde; while catalase, in the peroxisomes, requires hydrogen peroxide to oxidize alcohol [53]. Acetaldehyde, produced by alcohol oxidation through any of the mechanisms outlined above, is rapidly metabolized to acetate, to form acetate and NADH. The NADH is then oxidized in the mitochondria. Acetaldehyde has the capacity to form protein adducts in hepatocytes, and this has been proposed as an initiator of pathological processes. Structural mitochondrial alterations caused by acetaldehyde lead to functional impairment, including to decreased ATP generation via the respiratory chain, the production of ROS and a decrease in acetaldehyde dehydrogenase activity (an enzyme located in mitochondria that is responsible for the metabolism of acetaldehyde to acetate) [53][54]. These metabolic changes are exacerbated by impaired β-oxidation and decreased very-low-density lipoprotein secretion, promoting lipid infiltration of the hepatocytes. Such irreversible metabolic changes in liver cells impair the ability of the liver to recover.
Violation of the recovery capacity of liver in ALD is also reflected in a decrease in the proliferative activity of hepatocytes. Dippold, R. P. et al. showed that after PH, DNA synthesis was, at 24 h, already drastically inhibited in the livers of ethanol-fed rats compared with pair-fed rats. Delay in proliferation in the ethanol-treated liver is associated with a lack of induction of cell cycle genes after PH [55]. There was also impairment of normal miRNA signaling during the regeneration process. At a cellular level, chronic alcohol consumption induces senescent replication of hepatocytes [56]. Evidence suggests that hepatocyte-specific inhibition of miR-122 is associated with features of ALD in mice, and a combination of alcohol feeding and miR-122 inhibition accelerates alcohol-induced liver injury, steatosis, inflammation and fibrosis [53].
When the disease passes into a chronic form, inflammation is the main factor involved in the progression of ALD. While both ethanol and acetaldehyde are direct hepatotoxins, excessive ROS production and the subsequent production of inflammatory cytokines can promote alcohol-induced liver injury and inflammation. In addition, ethanol and acetaldehyde directly injure hepatocyte mitochondria, upregulating mitochondrial ROS production and further promoting liver injury and inflammation [57]. Damage-associated molecular patterns are evident after cell death—mainly necrosis—and trigger macrophage and neutrophil activation, fibrogenesis and hepatic regeneration [57].
6. Conclusions
This text has described the main known mechanisms that reduce the regenerative potential of liver with the most common liver pathologies. The key aspects are impaired hepatocyte metabolism and lack of signaling for the regenerative process. Despite the different origins of the diseases described, the main metabolic mechanisms are similar, the key factor being oxidative stress, which leads to disruption of the integrity of the cell structure or the mitochondrial membrane. In turn, disruption of mitochondrial integrity triggers a vicious cycle leading to mitochondrial dysfunction and, ultimately, to death of the liver cells. Next, two scenarios follow. In the first case, activation of compensatory pathological restoration of the liver tissue occurs, due to both the proliferation of hepatocytes (or, in some cases, of progenitor liver cells) and the growth of the ECM (fibrogenesis). These events lead to the development of fibrosis or even neoplasia. In the second case, the liver’s ability to recover is irreversibly compromised, resulting in liver failure, a serious condition with a high lethal risk. Understanding the mechanisms that underlie not only the normal but also the violated regenerative processes could enhance therapeutic opportunities.
Due to the wide spread of concomitant hepatic pathologies, the question arises in the search for new approaches of how to ensure appropriate liver regeneration. The main challenge is the targeted delivery of regeneration-stimulating molecules to specific cells while maintaining the molecules’ bioactive properties. It is also important to develop modern methods for assessing the state of the liver. Furthermore, new clinical diagnostic methods need to be based not only on assessment of the liver volume and its tissue structure but also on the provision of predictive assessments of the regeneration potential of the liver remnant. It is evident that the development of new methods for assessing the regenerative potential of liver remnants and of strategies to stimulate such regeneration should not underestimate pathological changes at the level of cell metabolism.
Funding: The work was supported by the Russian Science Foundation Grant No. 19-15-00263.
This entry is adapted from the peer-reviewed paper 10.3390/ijms24119112
References
- Guglielmi, A.; Ruzzenente, A.; Conci, S.; Valdegamberi, A.; Iacono, C. How much remnant is enough in liver resection? Digest. Surg. 2012, 29, 6–17.
- Bhushan, B.; Walesky, C.; Manley, M.; Gallagher, T.; Borude, P.; Edwards, G.; Monga, P.S.; Apte, U. Pro-regenerative signaling after acetaminophen-induced acute liver injury in mice identified using a novel incremental dose model. Am. J. Pathol. 2014, 184, 3013–3025.
- Golse, N.; Bucur, P.O.; Adam, R.; Castaing, D.; Sa Cunha, A.; Vibert, E. New paradigms in post-hepatectomy liver failure. J. Gastrointest. Surg. 2013, 17, 593–605.
- Nilsson, H.; Karlgren, S.; Blomqvist, L.; Jonas, E. The inhomogeneous distribution of liver function: Possible impact on the prediction of post-operative remnant liver function. Hpb 2015, 17, 272–277.
- Truant, S.; Scatton, O.; Dokmak, S.; Regimbeau, J.M.; Lucidi, V.; Laurent, A.; Gauzolino, R.; Castro Benitez, C.; Pequignot, A.; Donckier, V. Associating liver partition and portal vein ligation for staged hepatectomy (ALPPS): Impact of the inter-stages course on morbi-mortality and implications for management. Eur. J. Surg. Oncol. 2015, 41, 674–682.
- Moris, D.; Vernadakis, S.; Papalampros, A.; Vailas, M.; Dimitrokallis, N.; Petrou, A.; Dimitroulis, D. Mechanistic insights of rapid liver regeneration after associating liver partition and portal vein ligation for stage hepatectomy. World J. Gastroenterol. 2016, 22, 7613.
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922.
- Peng, C.; Stewart, A.G.; Woodman, O.L.; Ritchie, R.H.; Qin, C.X. Non-alcoholic steatohepatitis: A review of its mechanism, models and medical treatments. Front. Pharmacol. 2020, 11, 603926.
- Hamano, M.; Ezaki, H.; Kiso, S.; Furuta, K.; Egawa, M.; Kizu, T.; Chatani, N.; Kamada, Y.; Yoshida, Y.; Takehara, T. Lipid overloading during liver regeneration causes delayed hepatocyte DNA replication by increasing ER stress in mice with simple hepatic steatosis. J. Gastroenterol. 2014, 49, 305–316.
- Allaire, M.; Gilgenkrantz, H. The aged liver: Beyond cellular senescence. Clin. Res. Hepatol. Gastroenterol. 2020, 44, 6–11.
- Schaub, J.R.; Malato, Y.; Gormond, C.; Willenbring, H. Evidence against a stem cell origin of new hepatocytes in a common mouse model of chronic liver injury. Cell Rep. 2014, 8, 933–939.
- Brunt, E.M. Nonalcoholic fatty liver disease and the ongoing role of liver biopsy evaluation. Hepatol. Commun. 2017, 1, 370–378.
- Veteläinen, R.; van Vliet, A.; Gouma, D.J.; van Gulik, T.M. Steatosis as a risk factor in liver surgery. Ann. Surg. 2007, 245, 20.
- De Meijer, V.E.; Kalish, B.T.; Puder, M.; IJzermans, J.N.M. Systematic review and meta-analysis of steatosis as a risk factor in major hepatic resection. J. Br. Surg. 2010, 97, 1331–1339.
- Kele, P.G.; van der Jagt, E.J.; Gouw, A.S.; Lisman, T.; Porte, R.J.; de Boer, M.T. The impact of hepatic steatosis on liver regeneration after partial hepatectomy. Liver Int. 2013, 33, 469–475.
- Chu, M.J.; Hickey, A.J.; Phillips, A.R.; Bartlett, A.S. The impact of hepatic steatosis on hepatic ischemia-reperfusion injury in experimental studies: A systematic review. BioMed Res. Int. 2013, 2013, 192029.
- Basaranoglu, M.; Neuschwander-Tetri, B.A. Nonalcoholic fatty liver disease: Clinical features and pathogenesis. Gastroenterol. Hepatol. 2006, 2, 282.
- Krawczyk, M.; Bonfrate, L.; Portincasa, P. Nonalcoholic fatty liver disease. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 695–708.
- Michalopoulos, G.K. Liver regeneration after partial hepatectomy: Critical analysis of mechanistic dilemmas. Am. J. Pathol. 2010, 176, 2–13.
- Hijona, E.; Hijona, L.; Arenas, J.I.; Bujanda, L. Inflammatory mediators of hepatic steatosis. Mediat. Inflamm. 2010, 2010, 837419.
- Cusi, K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: Pathophysiology and clinical implications. Gastroenterology 2012, 142, 711–725.
- Hirsova, P.; Ibrahim, S.H.; Gores, G.J.; Malhi, H. Lipotoxic lethal and sublethal stress signaling in hepatocytes: Relevance to NASH pathogenesis. J. Lipid Res. 2016, 57, 1758–1770.
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism 2016, 65, 1049–1061.
- Wei, Y.; Rector, R.S.; Thyfault, J.P.; Ibdah, J.A. Nonalcoholic fatty liver disease and mitochondrial dysfunction. World J. Gastroenterol. 2008, 14, 193.
- Van Zutphen, T.; Ciapaite, J.; Bloks, V.W.; Ackereley, C.; Gerding, A.; Jurdzinski, A.; Moraes, R.A.; Zhang, L.; Wolters, J.C.; Bischoff, R.; et al. Malnutrition-associated liver steatosis and ATP depletion is caused by peroxisomal and mitochondrial dysfunction. J. Hepatol. 2016, 65, 1198–1208.
- Ibdah, J.A.; Bennett, M.J.; Rinaldo, P.; Zhao, Y.; Gibson, B.; Sims, H.F.; Strauss, A.W. A fetal fatty-acid oxidation disorder as a cause of liver disease in pregnant women. N. Engl. J. Med. 1999, 340, 1723–1731.
- Cheng, Z.; Ristow, M. Mitochondria and metabolic homeostasis. Antioxid. Redox Signal. 2013, 19, 240–242.
- Koves, T.R.; Ussher, J.R.; Noland, R.C.; Slentz, D.; Mosedale, M.; Ilkayeva, O. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008, 7, 45–56.
- Nassir, F.; Rector, R.S.; Hammoud, G.M.; Ibdah, J.A. Pathogenesis and prevention of hepatic steatosis. Gastroenterol. Hepatol. 2015, 11, 167.
- Marsman, H.A.; De Graaf, W.; Heger, M.; Van Golen, R.F.; Ten Kate, F.J.W.; Bennink, R.; Van Gulik, T.M. Hepatic regeneration and functional recovery following partial liver resection in an experimental model of hepatic steatosis treated with omega-3 fatty acids. J. Br. Surg. 2013, 100, 674–683.
- Gargouri, M.; Magné, C.; El Feki, A. Hyperglycemia, oxidative stress, liver damage and dysfunction in alloxan-induced diabetic rat are prevented by Spirulina supplementation. Nutr. Res. 2016, 36, 1255–1268.
- Gilgenkrantz, H.; de l’Hortet, A.C. Understanding liver regeneration: From mechanisms to regenerative medicine. Am. J. Pathol. 2018, 188, 1316–1327.
- Sarin, S.K.; Choudhury, A. Acute-on-chronic liver failure: Terminology, mechanisms and management. Nat. Rev. Gastroenterol. Hepat. 2016, 13, 131–149.
- Lucchesi, A.N.; Freitas, N.T.D.; Cassettari, L.L.; Marques, S.F.G.; Spadella, C.T. Diabetes mellitus triggers oxidative stress in the liver of alloxan-treated rats: A mechanism for diabetic chronic liver disease. Acta Cir. Bras. 2013, 28, 502–508.
- Francés, D.E.; Ronco, M.T.; Ingaramo, P.I.; Monti, J.A.; Pisani, G.B.; Parody, J.; Pellegrino, J.; Carrillo, M.C.; Martín-Sanz, P.; Carnovale, C.E. Role of reactive oxygen species in the early stages of liver regeneration in streptozotocin-induced diabetic rats. Free Radic. Res. 2011, 45, 1143–1153.
- Han, Y.; Sun, T.; Han, Y.; Lin, L.; Liu, C.; Liu, J.; Yan, G.; Human, R.T. umbilical cord mesenchymal stem cells implantation accelerates cutaneous wound healing in diabetic rats via the Wnt signaling pathway. Eur. J. Med. Res. 2019, 24, 10.
- Huang, X.L.; He, Y.; Ji, L.L.; Wang, K.Y.; Wang, Y.L.; Chen, D.F.; Geng, Y.; OuYang, P.; Lai, W.M. Hepatoprotective potential of isoquercitrin against type 2 diabetes-induced hepatic injury in rats. Oncotarget 2017, 8, 101545.
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218.
- Karsdal, M.A.; Manon-Jensen, T.; Genovese, F.; Kristensen, J.H.; Nielsen, M.J.; Sand, J.M.; Hansen, N.U.B.; Bay-Jensen, A.C.; Bager, C.L.; Krag, A.; et al. Novel insights into the function and dynamics of extracellular matrix in liver fibrosis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2015, 308, G807–G830.
- Liang, S.; Kisseleva, T.; Brenner, D.A. The role of NADPH oxidases (NOXs) in liver fibrosis and the activation of myofibroblasts. Front. Physiol. 2016, 7, 17.
- Crespo Yanguas, S.; Cogliati, B.; Willebrords, J.; Maes, M.; Colle, I.; Van den Bossche, B.; Souza de Oliveira, C.P.M.; Andraus, W.; Alves, V.A.; Leclercq, I.; et al. Experimental models of liver fibrosis. Arch. Toxicol. 2016, 90, 1025–1048.
- Tanaka, M.; Miyajima, A. Liver regeneration and fibrosis after inflammation. Inflamm. Regen. 2016, 36, 19.
- Donne, R.; Saroul-Aïnama, M.; Cordier, P.; Celton-Morizur, S.; Desdouets, C. Polyploidy in liver development, homeostasis and disease. Nat. Rev. Gastroenterol. Hepat. 2020, 17, 391–405.
- Dewhurst, M.R.; Ow, J.R.; Zafer, G.; van Hul, N.K.; Wollmann, H.; Bisteau, X.; Brough, D.; Choi, H.; Kaldis, P. Loss of hepatocyte cell division leads to liver inflammation and fibrosis. PLoS Genet. 2020, 16, e1009084.
- Michalopoulos, G.K.; Bhushan, B. Liver regeneration: Biological and pathological mechanisms and implications. Nat. Rev. Gastroenterol. Hepat. 2021, 18, 40–55.
- Dahlke, M.H.; Popp, F.C.; Bahlmann, F.H.; Aselmann, H.; Jäger, M.D.; Neipp, M.; Piso, P.; Klempnauer, J.; Schlitt, H.J. Liver regeneration in a retrorsine/CCl4–induced acute liver failure model: Do bone marrow-derived cells contribute? J. Hepatol. 2003, 39, 365–373.
- Hafez, M.M.; Al-Shabanah, O.A.; Al-Harbi, N.O.; Al-Harbi, M.M.; Al-Rejaie, S.S.; Alsurayea, S.M.; Sayed-Ahmed, M.M. Association between paraoxonases gene expression and oxidative stress in hepatotoxicity induced by CCl4. Oxidative Med. Cell. Longev. 2014, 2014, 893212.
- Wang, S.; Pacher, P.; De Lisle, R.C.; Huang, H.; Ding, W.X. A mechanistic review of cell death in alcohol-induced liver injury. Alcohol. Clin. Exp. Res. 2016, 40, 1215–1223.
- Song, M.; Yi, X.; Chen, W.; Yuan, Y.; Zhang, X.; Li, J.; Tong, M.; Liu, G.; You, S.; Kong, X. Augmenter of liver regeneration (ALR) gene therapy attenuates CCl4-induced liver injury and fibrosis in rats. Biochem. Biophys. Res. Commun. 2011, 415, 152–156.
- Ulger, O.; Kubat, G.B.; Cicek, Z.; Celik, E.; Atalay, O.; Suvay, S.; Ozler, M. The effects of mitochondrial transplantation in acetaminophen-induced liver toxicity in rats. Life Sci. 2021, 279, 119669.
- Bhushan, B.; Apte, U. Liver regeneration after acetaminophen hepatotoxicity: Mechanisms and therapeutic opportunities. Am. J. Pathol. 2019, 189, 719–729.
- Bhushan, B.; Chavan, H.; Borude, P.; Xie, Y.; Du, K.; McGill, M.R.; Lebofsky, M.; Jaeschke, H.; Krishnamurthy, P.; Apte, U. Dual role of epidermal growth factor receptor in liver injury and regeneration after acetaminophen overdose in mice. Toxicol. Sci. 2017, 155, 363–378.
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Prim. 2018, 4, 16.
- Barr, T.; Helms, C.; Grant, K.; Messaoudi, I. Opposing effects of alcohol on the immune system. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2016, 65, 242–251.
- Dippold, R.P.; Vadigepalli, R.; Gonye, G.E.; Patra, B.; Hoek, J.B. Chronic Ethanol Feeding Alters miRNA Expression Dynamics During Liver Regeneration. Alcohol. Clin. Exp. Res. 2013, 37, E59–E69.
- Louvet, A. Mathurin, Alcoholic liver disease: Mechanisms of injury and targeted treatment. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 231–242.
- Ohashi, K.; Pimienta, M.; Seki, E. Alcoholic liver disease: A current molecular and clinical perspective. Liver Res. 2018, 2, 161–172.
This entry is offline, you can click here to edit this entry!