Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Neurosciences
Amyotrophic lateral sclerosis (ALS) is a devastating progressive neurodegenerative disorder characterized by selective loss of lower and upper motor neurons (MNs) in the brain and spinal cord, resulting in paralysis and eventually death due to respiratory insufficiency. Pathological protein aggregates are also a feature of ALS, and occur in the form of ubiquitinated inclusions in neurons and glia.
- amyotrophic lateral sclerosis
- aggregation
- neurodegenerative diseases
1. Pathological Protein Aggregation Involved in ALS
As previously mentioned, protein aggregates are a pathological feature of several neurodegenerative diseases, including extracellular plaques of Aβ and intracellular neurofibrillary aggregates of tau protein in AD, or Lewy bodies in PD [26]. Pathological protein aggregates are also a feature of ALS, and occur in the form of ubiquitinated inclusions in neurons and glia (Figure 1).
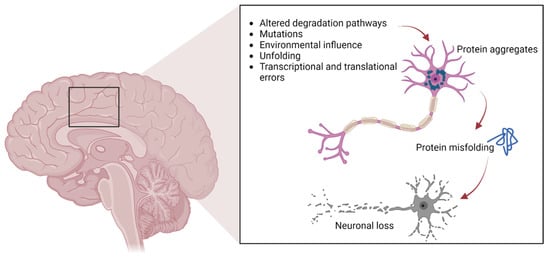
Figure 1. The pathways involved in protein aggregation in neuronal cells, leading to neuronal death typically found in neurodegenerative diseases.
Such inclusions contain different proteins, some of which may have an intrinsic tendency to aggregate following genetic mutations (SOD1, TDP-43, FUS), whereas others may simply be trapped in the aggregates [13,14,15,16,17,27]. In particular, cysteine-mediated aggregates of mutant SOD1 (mutSOD1) have been observed in ALS MNs, with the wt form of SOD1 also present, thus demonstrating the strong affinity and co-aggregation of the two forms of the protein [28]. It is widely believed that the toxic functions of mutSOD1 and other proteins typical of ALS are related to their tendency to aggregate. This hypothesis is supported by the presence of intracellular cytoplasmic inclusions, as well as mitochondrial inclusions rich in SOD1 identified in cell models and in animal spinal MNs, as well as in MNs of patients with ALS [27].
Numerous experimental theories have been proposed to explain how the aggregation of mutant proteins contributes to cellular toxicity in ALS patients. These suggestions have included the ability to sequester proteins necessary for the normal functioning of the MN [29], the ability to reduce the activity of the proteasome, which is essential for correct protein turnover, and the ability to inhibit the correct functioning of specific cellular organelles, such as mitochondria, by internal or external aggregation [30]. Cell degeneration depends on the sensitivity of MNs to the aggregation of proteins in the mutant form, supported by the observation thatTDP-43 and FUS also aggregate in patient tissues and ALS models in the same manner as SOD1 [31,32]. Indeed, anatomopathological observations of tissues derived from ALS patients show that these two proteins aggregate as cytoplasmic inclusions (positive for ubiquitin, but negative for SOD1) and that the mutations that affect them seem to increase the degree of aggregation [33]. Furthermore, it has been shown that the aggregation of FUS and TDP-43 is based on a conserved low-complexity domain (LCD), also called a prion-like domain (PrLD) [34], and that such aggregates can also sequester wt proteins and in their native form [10]. The LCD can mediate the liquid–liquid phase separation (LLPS) and therefore the formation of stress granules (SGs), which are cytoplasmic condensates lacking a membrane, composed primarily of RNA and RNA-binding proteins (RBPs), generated under unfavorable environmental conditions. SGs may serve to protect RNA from degradation by inhibiting the initiation of mRNA translation at the cellular level and starting the synthesis of cytoprotective proteins. They are intrinsically dynamic and dissolve quickly upon stress removal [35]. TDP-43 mislocalization and aggregation are observed in approximately 97% of all ALS cases, including all sALS cases, whereas SOD1 (2%) and FUS (1%) inclusions are associated with the remaining cases [36]. Not only are ubiquitin-immunoreactive inclusions most frequently reported in all forms of ALS, but the aggregates may also be reactive to p62, a protein that participates in sequestosome formation and autophagy [37].
To prevent the formation of protein aggregates, the cells are equipped with protein quality control systems, and the presence of chaperone proteins ensures that proteins in their native form fold correctly. These can intervene to avoid misfolding and therefore restore the proteins to the correct conformation shape: the UPS or the ALP. Dysfunction of these systems causes the formation of protein aggregates [38]. In this context, genetic screening has identified disease-associated mutations in many of the proteins identified within ALS inclusions [5]. These findings imply that aggregates are not merely a disease marker but are also strongly linked to the etiology and pathomechanisms of neurodegeneration.
2. SOD1
In 1993, Rosen et al. first described eleven disease-associated mutations in the SOD1 gene located on chromosome 21 [39], which encodes for the Cu/Zn superoxide dismutase, a cytoplasmic enzyme responsible for the catabolism of superoxide radicals to hydrogen peroxide and molecular oxygen [40]. SOD1 is ubiquitously expressed, highly conserved, and represents ~1% of all cytoplasmic proteins. It is a 32 kDa polypeptide of 153 amino acids, composed of a binding site with a zinc atom and another for a copper atom (Figure 2). This protein is predominantly localized in the cytoplasm of cells, although it is also apparently present in the intramembrane space of the mitochondria, nucleus, lysosomes, and peroxisomes [41]. Its role is to convert superoxide anions, which are toxic to the cell, into peroxides of hydrogen and oxygen [42]. SOD1 also exerts pro-oxidant activity including peroxidation with the production of hydroxyl radicals and nitration of tyrosines [43]. The ubiquitinated form of this protein is widely expressed and constitutes about 0.5–0.8% of soluble proteins in the human brain [44]. To date, over 200 mutations in SOD1 have been identified accounting for approximately 20% of fALS patients [45,46], characterized by inter-family and intra-family variability in phenotype with respect to severity of symptoms, age of onset, and disease duration [46]. The most common mutations are G93A, A4V, H46R, D90A, inherited as dominant traits, except for the latter that also shows a recessive inheritance pattern of transmission, but only in Scandinavian populations [32,47].
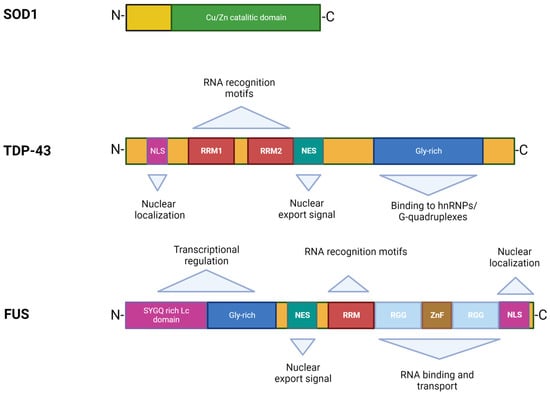
Figure 2. Structures and functional domains of SOD1, TDP-43, and FUS proteins.
The two principal critical features of SOD1 mediated cytotoxicity are misfolding and protein aggregation. This means that disease onset is driven by mutant protein that is synthesized inside MNs. MutSOD1 protein interacts specifically with neurofilament light chain mRNA and the dynein–dynactin complex, thus inducing cytoskeletal defects or altering axonal transport [47]. Furthermore, it has an increased tendency to form aggregate-prone monomers, and the degree of instability correlates inversely with survival time, suggesting that increased propensity to aggregation may be the unifying common denominator for different SOD1 mutations [47]. Researchers identified misfolded SOD1 in MNs in a subset of patients with sALS without SOD1 mutations, thus suggesting a role for wt SOD1 in sALS, possibly after secondary (oxidative) modification [48,49]. Finally, the aggregation and spread of mutant SOD1 has been demonstrated in cultured cells [50], and its seeding ability via a prion-like mechanism illustrated using spinal cord homogenate [51].
There are currently no explanations for how mutSOD1 causes disease. Initially it was hypothesized that the mutations impair the enzymatic activity of the protein, causing increased levels of reactive oxygen species (ROS) with consequent oxidative stress and death of neuronal cells [52]. More recent studies have shown that mutant protein forms maintain their catalytic activity intact with no apparent causal relationship between residual enzyme activity, clinical progression, and disease phenotype [53].
The mutSOD1 protein accumulates in the oligomeric form and produces cytoplasmic aggregates, which can cause neuronal cell death by sequestering other cytoplasmic proteins required for neuronal survival, blocking the UPS with consequent loss of chaperone proteins, destruction of mitochondria, and the blockade of cytoskeletal or axonal transport [54].
3. TDP-43
TDP-43 was first isolated in 1995, when researchers observed its ability to bind the transactivation response region (TAR) of HIV DNA, hence the name TAR DNA binding protein [55]. It was subsequently found in the human brain and in several cell culture systems [56]. TDP-43 is a highly conserved protein across different species and shows ubiquitous expression in humans and rodents with a predominant localization in the nucleus. This protein consists of 414 amino acids and has a molecular weight of 43 kDa, encoded by the TARDBP gene located on chromosome 1, a member of a heterogeneous family of proteins that bind RNA, known as hnRNP (heterogeneous ribonucleoprotein) [57]. From a structural point of view, the protein contains an N-terminal region, a nuclear localization signal (NLS), two RNA recognition motifs (RRM1 and RRM2) which exhibit an export signal nuclear (NES), and a C-terminal region comprising a glycine-rich LCD which mediates the interaction with other proteins belonging to the hnRNP family (Figure 2) [57]. Mutations affecting the TDP-43 protein represent about 5% of sALS and about 3% of fALS cases [58] as well as patients affected by frontotemporal dementia (FTD) [13,14]. The majority of TARDBP mutations are clustered mainly within the glycine-rich C terminal, and have a crucial role in nucleocytoplasmic shuttling, aggregation propensity, and protein–protein interaction [59]. The cellular function of TDP-43 remains unknown, but different studies have shown that this protein plays a fundamental role in a number of biological activities, including regulation of gene transcription, control of splicing processes, and maintaining the stability of mRNA [60]. Protein levels are strictly controlled through a self-regulation system, with particular involvement of the C-terminal region, further supporting the suggestion that mutations in this domain can interfere with the homeostatic control process by altering the recruitment of complexes required for self-regulation [61].
In pathological conditions, this protein tends to form ubiquitinated inclusions in the central nervous system (CNS), in particular the hippocampus, neocortex, and spinal cord. A further feature that distinguishes it from the wt form is its localization: insoluble protein aggregates tend to form in the cytoplasm in the neurons of patients with either pathology, resulting in a decrease of TDP-43 protein in the nucleus, in contrast to healthy subjects where it is expressed in the nucleus [13]. In addition to ubiquitination, hyperphosphorylation of TDP-43 is important for protein aggregation in ALS pathogenesis. In particular, in samples from ALS patients TDP-43 appears to be hyperphosphorylated at the C-terminal level, thus favoring its aggregation [62]. According to the findings of Braak and colleagues, pTDP-43 is widespread throughout the CNS, particularly in the agranular neocortex, and in spinal and bulbar MNs, where the presence of pTDP-43 makes it impossible to distinguish between the two types of MNs [63]. Furthermore, hyperphosphorylation of TDP-43 has been clearly linked to cell death in areas of the CNS, in relation to disease progression [64]. Another study also demonstrated that pTDP-43 aggregates led to the death of dopaminergic neurons in the substantia nigra [65].
Regardless of the presence of genetic mutations, the aberrant localization of TDP-43 in the cytoplasm of neurons in ALS patients appears to be linked to a pathogenetic mechanism associated with a loss of function in nuclear protein responsible for the regulation of mRNA transcription and splicing processes [60]. The formation of cellular inclusions of TDP-43 induces toxicity in the cell, and in this case the protein acquires a toxic function (gain of function) [66,67,68].
Furthermore, the accumulation of TDP-43 within ubiquitinated inclusions (UBIs) can lead to the altered regulation of genes or factors involved in the degradation processes of intracellular proteins, thus contributing to TDP-43 proteinopathy.
TDP-43 levels are strictly regulated by an intrinsic autoregulatory pathway, as demonstrated by observations that inactivation of one copy of the gene does not reduce protein mRNA levels in mice. Autoregulation is believed to be mediated by TDP-43 dependent splicing of an intron in the 3′ UTR region of its own mRNA, and splicing of this gene leads to unstable RNA which undergoes decay [69]. Furthermore, overexpression of wt human TDP-43 in neurons results in neurodegeneration accompanied by decreased locomotor activity, motor impairment, shorter lifespan, and MN loss [70].
TDP-43 is primarily cleaved by caspase 3 into two extremely aggregation-prone fragments with molecular weights of 25 kDa and 35 kDa, namely TDP-25 and TDP-35, respectively [71]. These fragments derive from the entire wt TDP-43 protein chain, induce greater toxicity, and contribute to the loss of function of TDP-43. Therefore, they must be efficiently cleared from cells to prevent their aggregation and the sequestration of other important neuronal components. The clearance of aberrant or misfolded proteins is mediated by the protein quality control system (PQC) [72]. This system is composed of chaperone and co-chaperone proteins that recognize and bind damaged proteins and direct them towards degradation processes.
As mentioned previously, the LCD sequence of TDP-43 controls its ability to undergo LLPS [73], leading to the formation of insoluble aggregates [74]. In particular, when aggregate-prone TDP-43 is more concentrated, the critical concentration for LLPS is exceeded, making it difficult to maintain protein homeostasis by preventing aggregation [62]. Further evidence of the importance of proper interactions between TDP-43 and LLPS comes from a recent study by Gao et al. performed on LLPS-deficient TDP-43 mice, showing that low levels of TDP-43 and LLPS led to the alteration of neuronal cells [75].
4. FUS
The discovery of TDP-43 mutations in ALS rapidly led to the identification of mutations in another RNA binding protein, namely FUS [16,17], accounting for 4–5% of fALS and 1% of sALS forms associated with young age at onset and short survival time [5,76]. The FUS gene is located on chromosome 16 and encodes for a 526-amino acid protein of 75 kDa in weight, showing a structure similar to that of TDP-43 (Figure 2) [77,78]. Although FUS was initially identified as a component of a fusion oncogene resulting from a chromosomal translocation observed in liposarcomas [60], this protein also plays a role in RNA processes [79]. FUS is widely expressed in most human tissues [80] and is primarily localized at the nucleus, although it shuttles to the cytoplasm to mediate a wide range of cellular processes including DNA repair, genomic stability, transcriptional regulation, splicing, transport, and maturation of mRNAs [60,81]. Specifically in the CNS, FUS regulates mRNA transport towards the dendrites and supports synaptic plasticity upon activation of glutamate receptors [82].
As with TARDBP, the majority of pathogenic mutations in FUS map to the C terminal within the NLS region of the protein that regulates interaction with transportin-1 [83], thereby interfering with the nuclear–cytoplasmic balance of FUS [78,82]. Accordingly, this protein exhibits mainly nuclear localization under healthy conditions, but abnormal cytoplasmic aggregates have been found in the brains and spinal cords of ALS patients with FUS mutations [16,57,82]. Therefore, the toxic effects of FUS seem to be related to its aberrant cytoplasmic localization that possibly disrupts nucleocytoplasmic transport [84]. This evidence is supported by a study in Drosophila where deletion of the nuclear export signal reduced the toxicity of mutant FUS [85].
Furthermore, authors have reported that FUS can undergo phase transition by LCD sequence [86] and formation of GSs [80], similar to TDP-43 [75]. Study in vitro demonstrated that ALS mutations may accelerate the kinetics of phase separation and exacerbate the transition of FUS from liquid to solid phase [86]. Further studies confirmed that an aberrant phase transition is reflected in the molecular mechanism underpinning ALS pathogenesis [87,88,89,90].
Interestingly, neuropathological examination of tissues from patients harboring FUS mutations showed increased cytoplasmic FUS staining, FUS-immunoreactive dystrophic neurites, and cytoplasmic inclusions in lower MNs [16,17]. These mislocalized immunoreactive FUS inclusions were strikingly non-reactive for TDP-43, suggesting that neurodegenerative processes driven by FUS are independent from TDP-43 mislocalization.
This entry is adapted from the peer-reviewed paper 10.3390/ijms24010704
This entry is offline, you can click here to edit this entry!