Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
Inflammasomes are important intracellular multiprotein signaling complexes that modulate the activation of caspase-1 and induce levels of the proinflammatory cytokines interleukin-1β (IL-1β) and IL-18 in response to pathogenic microorganisms and molecules that originated from host proteins. Inflammasomes play contradictory roles in the development of inflammation-induced cancers. Based on several findings, inflammasomes can initiate and promote carcinogenesis. On the contrary, inflammasomes also exhibit anticancer effects by triggering pyroptosis and immunoregulatory functions.
- cancer
- inflammation
- inflammasome
- pyroptosis
- colorectal cancer
1. Introduction
Inflammasomes are critical regulators of inflammation by stimulating the secretion of proinflammatory cytokines after sensing harmful pathogens and endogenous danger signals by innate immune system receptors (pattern-recognition receptor (PRR)) [1,2]. The tight regulation of inflammasomes is essential, as their deficient activation can lead to exacerbating and persistent pathogenic infections while upregulation can cause autoinflammatory disorders [3,4]. Therefore, understanding the structure of the inflammasome complex and activation/regulation mechanisms could help to modulate and manage different disorders.
Inflammasomes are multiprotein complexes that activate inflammatory caspases in response to pathogens or danger signals. The activation of caspases lead to the production of inflammatory cytokines (specifically, interleukin 1β (IL-1β) and interleukin 18 (IL-18)) and the induction of pyroptotic cell death [5]. IL-1β and IL-18 have a central role in inflammation and immunity, and it has been established that IL-1β/IL-18 activation by inflammasomes are considered the main factors in various inflammatory disorders [6,7]. Assembling this cytoplasmic complex is triggered by the activation of the PRR by pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs). Receptors that are able to assemble inflammasomes include leucine-rich repeat containing proteins (NLR) family members (like NLRC4, NLRP1 and NLRP3); proteins absent in melanoma 2 (AIM2) and pyrin (MEFV) [5]. NLRs have a nucleotide-binding and oligomerization domain (NACHT), which is located in the center and takes part in the oligomerization and dNTPase activity. NLRs also have either a caspase recruitment domain (CARD) or pyrin domain (PYD) and, in a few cases, a baculovirus IAP repeat (BIR) or a leucine-rich repeat (LRR). An apoptosis-associated speck-like protein containing a CARD (ASC) is an adaptor protein that acts as a bridge between upwards inflammasome components and caspase-1. The PYD and CARD domains are the main components of ASC. The interaction between the PYD domain of ASC and the PYD of the NLR leads to the aggregation of ASC molecules and formation of ASC filaments. On the other hand, the CARD domain of ASC interacts with the CARD of the Zymogen form of caspase-1. The Zymogen form of caspase-1 matures into caspase 1 through the proteolytic reaction. The interaction of PYD with ASC leads to the recruitment of the Zymogen form of caspase-1 and the activation of caspase-1 [8]. Upon activation, caspase-1, through its proteolytic cleavage properties, stimulates the maturation of the dominant proinflammatory precursor cytokines (IL-1β and IL-18) and releases active forms of these cytokines. Additionally, caspase-1 cleaves the substrate gasdermin D into an N-terminal fragment of gasdermin D that induces pyroptosis [9,10].
ASC has a dual role associated with cancer. It has been demonstrated that ASC expression is silenced via methylation, which inhibits tumor cell apoptosis. On the other hand, as mentioned earlier, ASC is also recognized as an inflammasome complex adaptor molecule, which mediates inflammatory cytokines production (such as IL-1β and IL-18), mediating tumor-promoting functions. Therefore, ASC may perform opposing functions, promoting tumor progression by increasing inflammatory cytokines production or tumor-suppressing by provoking tumor cell apoptosis [11].
In addition to the activation of caspases and proinflammatory cytokines, the activation of inflammasomes leads to the programmed cell death pyroptosis as a gasdermin-dependent form of cell death [12]. This is the term used to describe the release of cytoplasmic components in the extracellular space by the creation of membrane pores [13]. Pyroptosis, as an inflammatory type of programmed cell death, can protect against intracellular pathogens via removing intracellular replication niches and concurrently triggering an inflammatory response [14]. The phagocytes (dendritic cells, macrophages and neutrophils); CD4+ T cells; epithelial cells; endothelial cells; keratinocytes and neurons also undergo pyroptosis [15]. The PRRs that these cells express can recognize a broad spectrum of PAMPs and DAMPs upon microbial infection. The common PRRs include Toll-like receptors (TLRs) and NOD-like receptors (NLRs) [16]. PAMPs and DAMPs as the main stimuli trigger the formation of multiprotein complex inflammasomes, which later activate the caspases to induce pyroptosis. The inflammasome-mediated pyroptosis pathway may be canonical or noncanonical, with the prior applying caspase-1-activating inflammasomes and the end utilizing other caspases [17].
In pyroptosis, unlike apoptosis, a different set of caspases, such as caspase-1/4/5 in humans and caspase-11 in mice, are activated by the inflammasome [18]. These caspases will lead to the activation of several proinflammatory cytokines and the pore-forming protein gasdermins. The pores that are formed during pyroptosis will result in cell membrane rupture and cytokine release, as well as the release of various DAMPs such as DNA, HMGB-1 and ATP outside of the cell. DAMPs recruit immune cells and sustain the inflammatory cascade in the tissue [19,20].
The canonical inflammasome pathway is a two-step process, involving the priming and activation steps (signals 1 and 2). At first, signal 1 is provided by TNF-α and IL-1 or via the sensing of PAMPs and DAMPs by TLRs or NOD1/2, which activate NF-κB. NF-κB activation will lead to the induction of pro-IL-1β and pro-IL-18 expression. Additionally, priming prepares the inflammasomes for activation through another undiscovered mechanism. The second step is followed by PAMP and DAMP sensing by NLRs (NLRP3, NLRC4, etc.) or AIM2 and pyrin via mechanisms that are not fully understood [21]. The activation of these receptors increases the assembly of the NLRP3 inflammasome, caspase-1-mediated IL-1β and IL-18 release and, subsequently, pyroptosis. A sensor protein (PRRs), an adaptor (ASC) and an effector (caspase-1) are the main components of the canonical pathway [17].
Some NLRs, like NLRP3 and NLRC4, using ASC, will interact with the Zymogen form of caspase-1, while others like NLRP1 can directly interact with caspase-1. The Zymogen form of caspase-1 activation, and its cleavage, can catalyze the proteolytic cleavage and activation of IL-1β and IL-18. Furthermore, caspase-1 activation is likely to have a direct effect on the induction of pyroptotic cell death [21].
In addition, non-inflammasome-forming PRRs like TLRs and NOD1/NOD2 are also important in pyroptosis. These receptors, via the activation of NF-κB and MAPK-signaling pathways, will upregulate inflammatory cytokine expressions (IFN α/β,TNF, IL-12 and IL-6) [22,23]. These active inflammatory cytokines will be released from the host cells. Caspase-1 also cleaves the cytosolic gasdermin D (GSDMD) to generate an N-terminal domain (GSDMD-N) and a C-terminal domain (GSDMD-C) [24]. In normal conditions, the GSDMD-C auto-represses GSDMD-N cleavage to suppress cell lysis [25]. GSDMD-N via oligomerization forms pores in the plasma membrane with an inner diameter of 10-14 nm, which facilitates potassium efflux and the release of IL-1β, IL-18 and other cytosolic components into the extracellular area. This interrupts the cellular ionic gradient, leading to an increase in osmotic pressure and, subsequently, pyroptosis [24,25]. GSDMD-N restricts the damage to normal neighboring cells by inserting itself into the inner membrane [26].
The activation of caspase-4/5 in humans and caspase-11 in mice responds to the noncanonical inflammasome pathway, which is initiated by the binding of a lipopolysaccharide (LPS) of Gram-negative bacteria directly onto these caspases. The oligomerization and activation of these caspases cleaves GSDMD to release GSDMD-N, triggering pyroptosis. This effect is seen to increase with LPS binding to the caspases [27]. Furthermore, the influx of potassium ions upon membrane permeabilization also stimulates NLRP3 activation, resulting in NLRP3 inflammasome formation and the activation of caspase-1. These processes promote GSDMD cleaving and improve the maturation and release of the proinflammatory cytokines [17].
Previous studies have revealed that the high production of IL-1β and IL-18, through the dysregulation of inflammasome activity during the microbial invasion or intestinal inflammation, plays a critical role in the pathogenesis of inflammatory bowel diseases (IBD) and colorectal cancer. However, the exact role of the inflammasome in the progression of these disorders requires more investigation [28].
IBD (ulcerative colitis and Crohn’s disease) is at an enhanced risk of colorectal cancer (CRC) development. The disease duration, stage of disease, degree of mucosal inflammation and portion of the bowel, family history, the related primary sclerosing cholangitis and age at diagnosis are the main CRC risk factors [29,30]. The development of CRC is two to six times greater in IBD patients compared to the general population. The major factor of CRC development in IBD is dysplasia, which is a neoplastic intraepithelial transformation. The inflammation induces intestinal epithelial cell (IEC) apoptosis through tumor suppressor p53 pathways; impaired signaling by p53 may be an initial step of the dysplasia progression to cancer [30].
2. Inflammasomes and Inflammatory Bowel Disease
IBD is a chronic gastrointestinal inflammatory condition that is categorized into ulcerative colitis (UC) and Crohn’s disease (CD). IBD is the result of genetic susceptibility to the dysregulation of the immune response to bacterial antigens in the gut lumen under certain environmental factors [31,32]. The intestinal innate and adaptive immune systems, the integrity of the epithelial barrier, the balance of commensal microbiota (dysbiosis) and diet are the main factors of IBD pathogenesis [33]. The symptoms of UC and CD can be similar, such as abdominal cramps, fever, bowel diarrhea with hemorrhage and/or containing mucus; however, the location and pattern of inflammation will differ. The location of inflammation in CD may appear anywhere along the digestive tract from the mouth to the anus and affects all layers of the bowel walls. In contrast, the inner lining of the colon is the only section that is affected in UC and begins in the rectum [34,35]. One of the main characteristics of UC is crypt abscesses formed by neutrophil migration through the intestinal epithelium. Contrary to this, the formation of granulomas, fissures and fistulas is the main inflammatory characteristic of CD [34,35]. Studies examining the cytokines in IBD have demonstrated that inflammatory cytokines such as IL-1β and IL-18 are correlated with active IBD, and IL-18 gene polymorphisms are associated with CD [36,37,38]. More specifically, Th2-type cytokines are involved in the pathogenesis of UC, whereas CD is correlated to Th1 and Th17 cytokines [33,34,35].
CD4 helper T (Th) cells, upon activation, differentiate into two main effector subsets (Th1 or Th2). Th1 cells mediate cellular immunity, the defense against intracellular pathogens and the development of various types of immunopathological reactions by producing proinflammatory cytokines such as interferon-g (IFN-γ) and lymphotoxin-a (LT-α). In contrast, Th2 cells mediate humoral immune responses; the defense against intestinal nematodes and the development of atopic disorders by producing cytokines (IL-4, IL-5, IL-13, IL-9 and IL-10) that modulate the proliferation and antibody class-switching of B cells [39,40].
Recent evidence in candidate-gene approach studies suggest an association between CD and the NLRP3 gene. Genome-wide association studies (GWAS) have discovered that the genetic susceptible element of IBD includes more than 70 susceptibility loci for CD and 40 susceptibility loci for UC [41,42]. It is known that the innate immune system—specifically, inflammasomes—contributes to chronic inflammatory disorder pathologies, like IBD [43].
It has been shown that the various polymorphisms of the NLRP3 gene might result in a decrease in the expression of the NLRP3 inflammasome and, subsequently, influences IBD genetic susceptibility. Briefly, single nucleotide polymorphisms (SNPs) in the NLRP3 genes (rs10733113); the C10X allele in CARD8; the Q705K allele in NALP3; CARD8; IL-18 (rs1946518 A > C, rs360718 A > C and rs187238 G > C) and CARD15/NOD2 are CD-susceptible polymorphisms of the NLRP3 inflammasome. SNPs in the NLRP3 inflammasome (rs10925019 and rs10754558) are UC-susceptible polymorphisms of the NLRP3 inflammasome [44].
Notably, Villan et al. [45] discovered that the SNP rs10733113 in the NLRP3 gene loci is potently associated with CD disease susceptibility. However, another study [46] could not replicate this association in a UK Panel. It was revealed that the lack of function of the CARD8 mutation in the NLRP3 inflammasome contributes to CD development [47,48]. Furthermore, the C10X allele in CARD8 accompanied by an Q705K allele in NALP3, together with NOD2 wild-type alleles, results in CD susceptibility in Swedish men [49]. Moreover, some of the polymorphisms of the NLRP3 effector IL-18 (rs1946518 A > C, rs187238 G > C and rs360718 A > C) were reported to be correlated with enhanced CD susceptibility [50]. In Han Chinese, the NLRP3 SNPs rs10754558 and rs10925019 could participate UC susceptibility but not to CD [51]. Furthermore, rs10754558 polymorphisms with “GG” and “CG” genotypes have been significantly associated with UC in Iranian patients [52]. The association between some of the SNPs that affect the receptors downstream of NLRP3 include IL18R1, IL1RL1, IL1R1, IL1R2 and IL1RL2, and a susceptibility to IBD was shown in a recent GWAS meta-analysis [53]. Collectively, various studies have demonstrated that the NLRP3 inflammasome represents a significant role in colitis pathogenesis, although the results are still questionable.
The inflammasome components (caspase-1, ASC and NLPR3) participate in a cell-specific regulation of the inflammasome-induced response. Data from several sources have identified that different intestinal cell types (epithelial and hematopoietic cells) express components of inflammasomes to respond against commensal microbiota and various pathogens for preventing mucosal damage and/or cause systemic disease [43].
Inflammasome expression by intestinal epithelial cells (IEC) is anticipated to be critical for intestinal immune homeostasis, mucosal immune defense, inflammation and tumorigenesis [54]. Data extracted from purified IEC, in-situ detection and cell-specific ablation have exhibited an array of inflammasome component expressions within IEC, including NLRC4, NAIP, NLRP1, NLRP6, caspase-1, caspase-4/5 (human), caspase-11 (mouse), ASC, AIM2 and IL-18 [55,56]. It has been demonstrated that the deletion of inflammasome components is associated with an enhanced susceptibility to damage and infection. Nevertheless, inflammasomes are regulated differently in IEC compared to the classic hematopoietic cells due to the individual intestinal environment. Based on the studies, the IEC produces relatively less IL-1β [57,58] and constitutively expresses IL-18 [59]. The IEC has a diverse inflammasome composition and can also secrete other possible factors besides IL-18 upon inflammasome activation, such as prostaglandin production, which has been correlated with NLRC4 activation [60].
3. Inflammasomes Regulate Intestinal Inflammation
Proinflammatory cytokines play a central role in carcinogenesis following chronic inflammation. In this case, IL-1 activates innate and adaptive immune cells and triggers these cells to produce additional inflammatory cytokines, chemokines and chemical mediators for infection and the injury response. Cell proliferation, repair and the healing process are the other roles of IL-1 [6]. Activated IL-1β and IL-18 induce the inflammatory pathway in myeloid and non-myeloid cell types [61,62]. Therefore, the use of chemical inhibitors of caspase-1, IL-1β and IL-18 can help to understand the exact role of these molecules in colitis pathogenesis [63,64,65,66].
On the other hand, a non-pyroptotic function of GSDMD in controlling the release of small extracellular vesicles (sEVs) contained from intestinal epithelial cells (IECs) in response to the activation of the caspase-8 inflammasome was described by Bulek et al. (2020). This study reported that GSDMD, which is activated by the caspase-8 inflammasome accompanied by Cdc37/Hsp90, recruits NEDD4 as an E3 ligase to catalyze pro–IL-1β polyubiquitination, assisting as a signal for cargo loading into secretory vesicles. It has been reported that IBD patients and those with experimental colitis showed elevated epithelial-derived GSDMD expression. Bulek et al. (2020) also showed that GSDMD deficiency considerably decreases the severity of the disease, relating to the GSDMD-mediated release of IL-1β sEVs in the intestinal inflammation pathogenesis, as observed in IBD [67].
4. Inflammasomes Protect the Integrity of the Intestinal Epithelial Barrier
In this section, we focus on the function of inflammasomes in the protection against colitis and colorectal cancer. Based on recent evidence, NLRP3, NLRP6 and caspase-1-deficient mice were significantly more sensitive to dextran-FITC, a polysaccharide that is toxic to the epithelium of the colon, than wild-type mice [85,87,92]. In fact, provoking a cytotoxic attack (using a compound like dextran-FITC, as seen in the last studies) to the intestinal epithelium has been seen to induce repair responses identified by enhanced stem cell division at the base of the crypt to renew the injured enterocytes [93]. In this regard, IL-1β and IL-18 also participate in repairing the damaged intestinal epithelium [94]. In order to confirm these findings, Zaki et al. (2010) used NLRP3 and caspase-1-deficient mice treated with dextran-FITC. The authors reported that the wild-type and deficient mice all increased the epithelial cell proliferation during acute colitis; however, when comparing the groups, the deficient mice presented with a decrease in proliferation when compared to the wild-type. This indicates that inflammasomes are required for repairing damaged tissue [87,92].
Additionally, inflammasome activation in myeloid cells has several outcomes. NLRP3 is originally expressed in macrophages, monocytes, dendritic cells (DC), granulocytes, osteoblasts and epithelial cells. The expression of NLRP3 in myeloid cells is profoundly inducible. Myeloid cells like macrophages, DC and granulocytes, in addition to lymphoid cells (T cells), express NLRP6. Pending development, NLRP6 is activated by PPAR-γ in the intestinal epithelium. Studies have discovered that NLRP6 plays a role in protecting against experimental colitis and colitis-associated cancer and monocytes [95]. The evaluation of antitumor immunity via DC vaccination demonstrated that the expression of NLRP3 in the tumor microenvironment promoted the migration of tumor-associated myeloid-derived suppressor cells (MDSCs) to the site of the tumor. Furthermore, the increased expression of NLRP-3 in MDSCs following this treatment was able to overcome the antitumor immunity [95]. A related study reported that Nlrp3-deficient mice also have fewer MDSCs accumulating at the region of the tumor and improved survival upon DC vaccination [96].
Bone marrow chimera studies have confirmed the function of inflammasomes in maintaining the integrity of the epithelial barrier, as well as protection against colitis and colorectal tumorigenesis. These studies have shown that acute colitis could be prevented by inflammasome activation in nonhematopoietic cells [84,92]. On the other hand, inflammasome activity in myeloid cells is critical for the suppression of polyp formation during chronic inflammation. This was reported by Allen et al. (2010), who used the AOM plus DSS tumorigenesis model and found that mice lacking caspase-1 and ASC had an increase in polyp formation, morbidity and disease outcome [84,92]. From these results, it can be deduced that intestinal epithelial cells activate inflammasomes during the acute phase of colon inflammation to repair the epithelial barrier and maintain the homeostasis of the gut epithelium. Myeloid cells may suppress the production of carcinogenic factors, which can provide a susceptible tumor microenvironment during the chronic phase of inflammation. Therefore, inflammasome activation in different cells and tissues could modulate inflammation and take part in tissue repair to prevent carcinogenic development in various stages of the disease [97].
In addition, other components of inflammasomes have recently been investigated, such as the NLR family of apoptosis inhibitory proteins (NAIP) (NAIP1, 2, 5 and 6 in mice and hNAIP in humans) and NLRC4. Recent studies have discovered that NAIPs and NLRC4 in intestinal epithelial cells respond to several stimuli, such as enteropathogenic bacteria invasion [98,99] and carcinogen exposure [91,100]. The main role of NAIPs is recognizing Gram-negative bacteria components such as flagella and type-three secretion systems (TTSS) and activate the NLRC4 inflammasome [101]. There is a strong correlation between NLRC4 and the function of phagocyte cells in order to eliminate the mucosal pathogen loads in the intestine lamina propria and other areas, the restriction of adverse pathology, systemic bacterial transmission and intestinal tumorigenesis [102,103,104]. Furthermore, NLRC4 has a protective role against acute mucosal infection by enteric bacteria like Salmonella typhimurium and Citrobacter rodentium [98,99]. NLRC4, through recruiting and activating the zymogen form of caspase-1, led to pro-IL-18 maturation [105]. IL-18 is a proinflammatory mediator that regulates proinflammatory responses in intestinal diseases, and its production may be a principal etiological factor for IBD patients [106,107].
5. NLRP6 Inflammasomes Maintain the Microbiota Balance
Intestinal epithelial cells are also involved in the maintenance of gut microbiota and modulation of the nutrient requirements for commensal organisms that secrete mucus and antimicrobial peptides to limit pathogen colonization. The dysregulation of this balance leads to dysbiosis, the outgrowth of “pathobionts” and increases the predisposition of a disease [113]. It is suggested that NLRP6, as one of the inflammasome components, is involved in maintaining the healthy microbiota in intestinal epithelial cells. It is likely that NLRP6, through an interaction with ASC, forms a canonical inflammasome [114]. NLRP6 is predominantly expressed in intestinal epithelial and goblet cells throughout the small and large intestines and nonepithelial cells of the gut mucosa (myofibroblasts and myeloid cells) [92,98,115,116,117].
The previously mentioned studies reported that NLRP6 deficiency leads to exacerbated DSS-induced colitis, as well as tumor progression in response to AOM/DSS. However, these studies could not explain the exact mechanism of NLRP6 for these phenotypes [92,115,116]. Other studies in this field have described that the protective effect of NLRP6 could be via the induction of IL-18 production by intestinal epithelial cells [115], hematopoietic cells [92] or lamina propria myofibroblasts by NLRP6 having an effect on intestinal epithelial cell proliferation and self-renewal [116]. Interestingly, colitis and carcinogenesis susceptibility with NLRP6, ASC or IL-18-deficient mice could be transferred to naive wild-type mice by cross-fostering and cohousing the mice together, leading to a dysbiotic gut microbiota [115,118]. However, the mechanisms of NLRP6 signaling that manage the gut microbiota conditions and inhibit the pathological drift are poorly understood. One study revealed that Nlrp6−/− mice have central defects in goblet cell granule release and a compromised mucus layer that correlates to autophagy defects [117]. Therefore, there is evidence that NLRP6 serves to improve the secretory activity of intestinal epithelial cells and mediates the release of IL-18 through other cell types.
6. Inflammasome and Intestinal Tumorigenesis
This section will review the latest studies that examine the correlation between inflammasomes and intestinal tumorigenesis and further discuss the contradictory roles of the inflammasome.
Colorectal cancer develops when tumor cells succeed to escape from both cell-intrinsic and cell-extrinsic cancer-inhibited mechanisms [119,120]. Several cancers prevent the antitumor immune response through the induction of low levels of inflammation, and hence, chronic inflammation could induce the development of tumor cells. The innate immune system prime adaptive immunity induces an inflammatory response that could eradicate tumor cells. IBD, as an example of an inflammatory disease, could progress to colorectal cancer [121,122].
Inflammasomes, as the main participants of inflammation, were considered to be associated with the modulation and progression of tumors. The remarkable epithelial responses, such as the proliferation of intestinal epithelial cells and tissue repair; chronic immune inflammatory responses, such as increased levels of cytokines, chemokines and ROS production and alterations in the intestinal microbiota content, were associated with colorectal tumor progression [121,123]. The main stimuli of inflammasome activation and its role in the IBD progression to CRC are demonstrated in Figure 1.
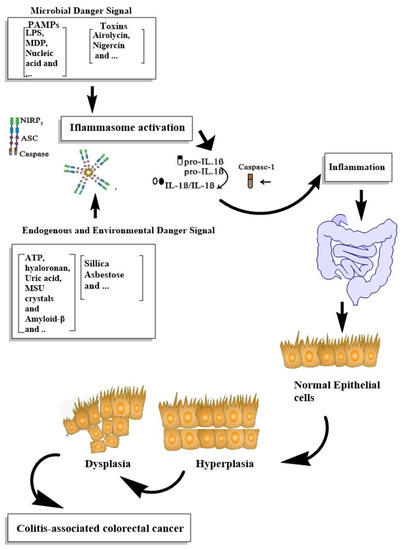
Figure 1. Summarizing the roles of inflammasome activation in IBD progression to CRC.
This entry is adapted from the peer-reviewed paper 10.3390/cells10092172
This entry is offline, you can click here to edit this entry!