Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Antibiotics have played a crucial role in the reduction in the incidence of tuberculosis (TB) globally as evidenced by the fact that before the mid-20th century, the mortality rate within five years of the onset of the disease was 50%. The use of antibiotics has eliminated TB as a devastating disease, but the challenge of resistance to anti-TB drugs, which had already been described at the time of the introduction of streptomycin, has become a major global issue in disease management.
- M.tb pathogenesis
- MDR-TB
- XDR-TB
- drug tolerance
1. Epidemiology of Tuberculosis
Although tuberculosis (TB) has been around for thousands of years, it is still one of the world’s biggest killers [1]. For centuries, tuberculosis has been a major killer of people living in poverty, overcrowding, and malnutrition, but in recent years the threat posed by this infectious disease has faded from public awareness [2]. To understand the reasons for the reduced focus on this infectious disease, it is important to look at the global trend in TB incidence over recent decades. By the mid-20th century, TB incidence and mortality had fallen in developed countries as a result of a better understanding of the disease and improvements in living conditions [3]. In addition, since the 1940s, the discovery of effective anti-tuberculosis drugs, which can significantly reduce the mortality rate due to this infection, has greatly accelerated this belief [4]. However, drug-resistant strains of TB began to emerge in the 1980s, leading the World Health Organization (WHO) to declare the problem a global health emergency in 1993 [5]. In addition, as a result of impaired cell-mediated immunity in HIV-infected patients, the incidence of TB began to rise again after decades of decline in the high-burden countries, such as areas of Sub-Saharan Africa [6]. Between 1995 and 2013, the detection rate of TB cases increased from 46% to 64% [7]. Since 1997, the WHO has published an annual Global Tuberculosis (TB) Report, which assesses the global TB situation and summarizes progress in the prevention, diagnosis, and treatment of the disease at all levels (national, regional and global) [8]. South-East Asia (43%), Africa (25%), and the Western Pacific (18%) have the highest proportion of TB cases. Smaller percentages occur in the Americas and Europe [9]. A major challenge in reducing tuberculosis mortality rates in developing countries is the difficulty in accessing health services, obtaining a diagnosis, and adhering to treatment regimes. Furthermore, diagnosis in these countries is often based on relatively insensitive methods, which are unable to detect drug-resistant strains. HIV has been the most important factor in the resurgence of tuberculosis (TB), particularly in Africa, and TB is a leading cause of death among people living with HIV worldwide (at least one-third of HIV-infected people also have TB) [10].
As part of its efforts to control TB, the WHO adopted the “Strategy to End TB” in 2014, with the aim of ending TB as a global public health threat by 2035 [11]. The End TB Strategy has had a significant impact, as shown by official data indicating that TB incidence and mortality have decreased by at least 20% and 35%, respectively, compared to the rates in 2015. [8]. There has also been an estimate of 49 million lives saved by 2015 [12]. However, despite the progress made, the decline in incidence has been disappointing and TB remains a major cause of morbidity and mortality in many countries [13]. The high number of TB deaths can be attributed to several factors: (1) one in three people with TB is not known to the health system and is therefore not treated; (2) treatment success rates remain too low in high-burden countries; and (3) rates of multidrug-resistant tuberculosis (MDR-TB), defined as resistance to the two main TB drugs, isoniazid and rifampicin, are increasing worldwide, with the emergence of TB caused by strains resistant to all current drugs [13][14]. The pillars of the WHO Strategy to End TB include three main lines of action to interrupt the trajectory of the TB epidemic: (1) integrated, patient-centered care and prevention; (2) bold policies and support systems; (3) intensification of research and innovation [8]. Although significant progress has been made in reducing the burden of TB, the rate of reduction remains slow and projections suggest that the global burden of TB as a global public health threat may not be eliminated by 2035, as envisaged in the Strategy to End TB [13]. According to the WHO, tuberculosis is the leading cause of death from infectious diseases worldwide, with about ten million new cases and 1.8 million deaths each year [12][15]. In 2021, there has been a decline in the diagnosis of TB and access to TB treatment as a result of the well-known COVID-19 pandemic [16][17]. The WHO estimates that 10.6 million people worldwide contracted TB in 2021, an increase of 4.5 per cent compared to 2020, causing 1.6 million deaths (including 187,000 HIV-positive people) and reversing years of global progress in reducing deaths from the disease [18]. MDR-TB is one of the leading causes of death due to antimicrobial resistance, accounting for one in three antimicrobial resistance-related deaths [19]. The number of MDR-TB infections is particularly worrying in China, India, and the Russian Federation [19][20].
2. Insights into Pathogenesis of Tuberculosis
Tuberculosis is an infectious bacterial disease caused by Mycobacterium tuberculosis (M.tb), which is airborne and most commonly affects the lungs (known as pulmonary TB), but can also spread to other parts of the body (known as extrapulmonary TB) [21]. As M.tb is essentially only found in humans (there is no animal reservoir for it), it has evolved to persist in humans for long time and only a fraction of people infected will develop active tuberculosis (about one quarter of the world’s population is latently infected with M.tb according to the WHO 2022 report) [15][22]. After initial infection, about 90% of infected people do not develop active disease, and M.tb can persist in the body for years (even a lifetime) without causing disease [22]. People with latent TB infection have no symptoms, are not contagious, and cannot spread TB to others. However, without treatment, the dormant mycobacteria can wake up and develop TB disease (active TB) in about 5% to 10% of infected people at some point in their lives [23]. The estimated lifetime risk of TB reactivation is much higher in immunocompromised patients, particularly those co-infected with HIV [24]. Tuberculosis is spread from person to person by aerosol droplets containing M. tuberculosis that are expelled from infected people when they cough, sneeze, or talk [25]. These tiny particles (≤5 microns in diameter), known as droplet nuclei, can remain suspended in the air for several hours in some conditions and can be transported more than 1 m. Inhaled infectious droplets travel through the respiratory tract and reach the alveoli of the lungs, where the tubercle bacilli are taken up by alveolar macrophages (AMs) of the host’s innate immune system [26]. Whether infection results in bacterial eradication, containment, asymptomatic infection, or active disease depends on the initial interaction between bacilli and AMs [26]. Thus, not all people exposed to an infectious TB patient will become infected with M. tuberculosis. The likelihood of TB transmission depends on several factors, the most important of which are: (1) the inhaled dose of infectious particles, which in turn depends on the bacillary load in the sputum of the patient with active TB; (2) the environment in which the exposure occurred (e.g., unventilated rooms increase the risk of droplet transmission); (3) the proximity of the individual to an infectious TB patient; (4) the duration of exposure (people in close contact with TB patients increase the risk of droplet transmission) [26][27][28]. If the macrophages fail to kill the bacilli, infected AMs migrate from the alveolar space into the lung interstitium, where the bacilli infect other cells such as DCs and different macrophage populations (Figure 1) [28]. The spread of bacilli from the site of infection is based on their ability to convert these antimicrobial cells into a permissive cellular niche [29]. At this stage, the bacteria can spread to any part of the body (e.g., lymph nodes, lungs, spine, bones, or kidneys) via the lymphatic and hematogenous pathways [30]. Numerous previous studies have shown that despite their host-protective role, AMs serve as a niche not only for M.tb growth, but also for facilitating the translocation of bacilli from the alveolar space into the interstitium prior to the arrival of recruited myeloid cells [31][32][33]. These studies clearly suggest that in the M.tb-infected lung, at least two macrophage subtypes are recruited to the site of infection and that M.tb has evolved several mechanisms that allow it to exploit the heterogeneity and plasticity of macrophages for productive infection and spread [33]. The M1 (pro-inflammatory)/M2 (anti-inflammatory) polarization of macrophages plays a crucial role in how TB infection progresses or regresses as a result of the responses they exert [33]. Moreover, recent studies have shown that at least four different subsets of macrophages which do not exhibit typical characteristics of either the M1 or M2 sublineages are involved as M.tb-permissive and M.tb-restrictive macrophage subsets [31]. Some macrophages can control infection more effectively than other cells by using anti-microbial mechanisms including phagolysosomal fusion, autophagy, and oxidative stress [29]. M.tb survives in macrophages by inhibiting phagosome maturation and phagolysosome fusion. In addition, two different forms of macrophage cell death have been described following M.tb infection: necrosis (a form of death that results in cell lysis) and apoptosis (a form of cell death that leaves the cell membrane intact) [34]. While apoptosis of infected macrophages allows bacterial replication to be controlled and is subsequently associated with reduced pathogen viability, necrosis represents a mechanism that allows bacteria to evade host defenses and spread [29][35]. Infected macrophages secrete chemokines and cytokines that activate neutrophils, which in turn release reactive oxygen species (ROS) and neutrophil extracellular traps (NETs) to kill M. tuberculosis [36]. To establish infection, M. tuberculosis inhibits ROS production by neutrophils which act as a niche for M. tuberculosis replication [36]. At the same time, the bacilli activate a cascade of immune responses, recruiting DCs that phagocytize and transport M.tb to the draining lymph nodes to activate the T-cell-mediated immune response [37]. Acquired cell-mediated immunity develops within 2–10 weeks of infection by stopping the multiplication of bacteria and preventing their further spread [38]. Immune cells, first infected macrophages, and neutrophils, then T and B lymphocytes, sequester M.tb in a granulomatous structure [31]. Bacterial control is established and the inflammatory response is reduced, leading to latent TB infection (LTBI) [39]. During this phase (known as primary infection), specific immunity develops, a positive skin reaction to tuberculin or an interferon-gamma release test is observed, but there are no clinical signs of TB, no culturable bacilli, and no manifestations of the disease [39]. Patients are infected with M.tb but do not have TB disease. In summary, primary infection can have several outcomes: (1) it can be eliminated by the host immune system; (2) it can progress to active disease, manifesting in less than 10% of infected individuals within 1–2 years of infection (more common in individuals co-infected with HIV or in the presence of other risk factors such as diabetes, obesity, and alcoholism); (3) it can be contained as a latent infection due to the ability of mycobacteria to enter a non-replicating persistent state in which they are resistant to therapy [40]. An effective adaptive immune response is required for the formation of granulomas, which are the result of the initial aggregation of macrophages (Figure 1) [41]. There are three main types of granulomas, representing different stages of a continuum: solid, M.tb-containing granulomas; necrotic granulomas, typical of early stages of active TB; and caseous granulomas, in late stages of TB [40]. The solid granuloma is composed of different macrophage morphotypes (epithelioid macrophages, foamy macrophages, multinucleated giant cells) and DCs, which form the central scaffold around which other cell populations, such as B and T lymphocytes, are arranged in concentric layers. Solid granulomas predominate in LTBI; these structures prevent the pathogen from spreading throughout the organism, but also allow M.tb to survive for decades by remaining in a slowly replicating state [42]. As the disease progresses, the granuloma undergoes a series of morphological changes due to the differentiation of macrophages first into epithelioid cells and then into multinucleated giant cells. The core, which consists of cell debris resulting from the necrotic lysis of host immune cells, becomes increasingly necrotic, often hypoxic, and forms a cheese-like structure known as a caseous granuloma (Figure 1) [42]. In the late stages of the disease, macrophages can transform into foam cells, either as a result of lipid droplet accumulation caused by dysregulation of host lipid metabolism or the deposition of mycolic acids [43]. Mature granulomas are dense aggregates of macrophages surrounded by an outer sheath of infiltrating lymphocytes, dominated by T and B cells. In summary, in the presence of an effective adaptive immune response, granulomas control and even sterilize infection by sclerosing and calcifying (solid granuloma) [28]. Conversely, a weak immune response results in the formation of a caseous granuloma, which acts as a reservoir, storing and harboring tubercle bacilli that, when the caseous core softens, cavitates and releases the bacilli, spreading not only to other organs but also to other people. This initiates the symptomatic phase of the disease, leading to active TB (Figure 1) [44]. Several virulence genes of Mycobacterium tuberculosis, which are crucial for pathogen survival and play a crucial role in its pathogenicity, have been identified [45]. Based on their function, these virulence factors can be divided into two primary groups. The first group includes virulence genes that encode enzymes involved in lipid pathways and proteins of signal transduction systems. The second category comprises virulence genes that facilitate the survival of mycobacteria in the hostile environment of host macrophages. An in-depth description of the mycobacterial virulence determinants has been reported in previous extensive reviews [45][46][47].
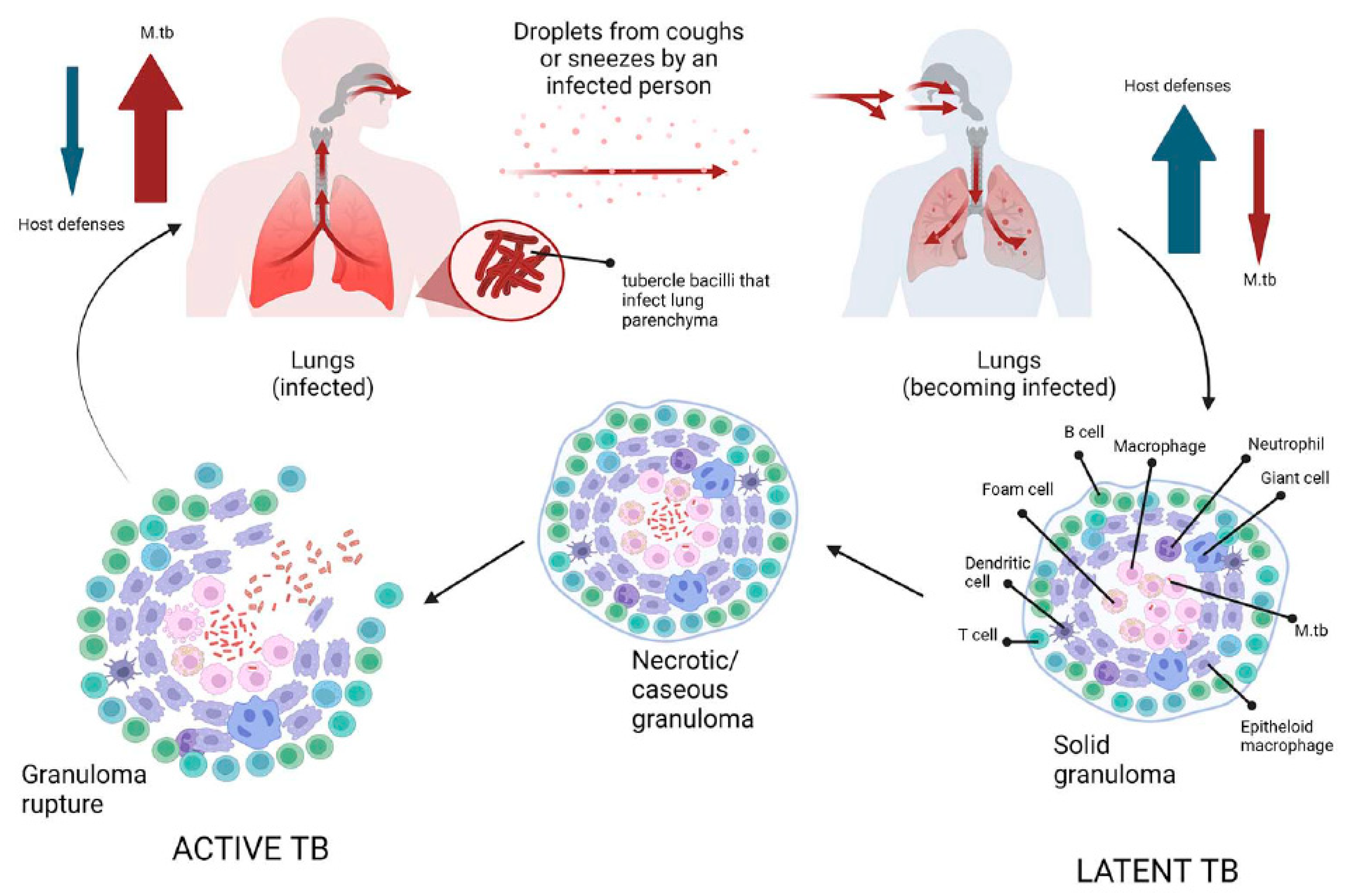
Figure 1. Schematic representation of tuberculosis pathogenesis. Infection with Mycobacterium tuberculosis occurs when aerosol droplets containing tubercle bacilli from an actively infected person reach the alveoli of an uninfected contact. In immunocompetent individuals, the host immune system fights infection by continuously recruiting and accumulating different morphotypes of macrophages, dendritic cells, B and T cells to form solid granulomas. This structure contains the bacilli, making healthy individuals latently infected (latent tuberculosis). As the disease advances, the granulomas undergo necrotic lysis of the immune cells, resulting in changes, and the center of the granuloma becomes caseous, which can lead to cavities in the lungs in the later stages of TB. If, for some reason, the host’s immune system fails to control the infection (such as in HIV or diabetes), the tubercle bacilli become active again, multiply, break out of the granuloma, and spread to other people, initiating the symptomatic phase of the disease (active TB).
3. Treatment of LTBI and TB Disease
Although many people can be infected with TB, not all will develop the disease. Depending on whether the host’s immune system is able to fight the bacteria and prevent its growth, there are two TB-related conditions: latent TB infection and TB disease [26]. If the immune system fails to stop the TB bacteria from growing, they will start to multiply in the body and cause TB disease [48]. Because not all people exposed to Mycobacterium tuberculosis develop latent tuberculosis, and even people with latent tuberculosis have no symptoms and are not infectious, there are two main ways of diagnosing LTBI: the historical tuberculin skin test and interferon gamma release assay test [22]. Neither of these tests is able to distinguish between an active and a latent TB infection [22]. International guidelines recommend testing for latent TB only in people at high risk of TB infection, such as those who work in hospitals or have other medical conditions (e.g., insulin-dependent diabetes) or a weakened immune system (e.g., HIV infection) [6]. The current recommended treatment regimen for latent TB is shorter than that for active TB and includes a short course of rifamycin therapy of three to four months (previously, it was nine months of isoniazid monotherapy) [49]. For people who cannot take a rifamycin-based regimen because of drug intolerance, the CDC recommended treatments include: (a) six months (for HIV-negative adults and children) or nine months of daily isoniazid; (b) three months of once-weekly isoniazid plus rifapentine [50]. Short-course regimens have both higher completion rates and a lower risk of hepatotoxicity than prolonged isoniazid monotherapy [51]. It is important to note that if treatment is not taken regularly or stopped too early, the bacteria can grow again and become resistant to the drugs. Unlike latent TB, the currently recommended treatment for any form of active TB (as mentioned above, TB can affect other parts of the body besides the lungs, including the lymph nodes, various organs, bones and joints, and even the brain) is the administration of long-term antibiotic therapy [51]. The most common regimens for active TB disease, according to recent WHO recommendations, involve the administration of more than one antibiotic over a period of four to nine months to ensure that all slow-growing TB bacteria are killed [52]. TB drugs fall into two categories: drugs for drug-susceptible TB (DS-TB), also known as ‘first-line TB drugs’ and drugs for drug-resistant TB (DR-TB), also called ‘second-line TB drugs’ [53]. The most common regimen for active DS-TB is isoniazid (INH) in combination with three other drugs: rifampicin (RIF), pyrazinamide (PZA), and ethambutol (EMB) (Figure 2). It is possible to recover from active TB, but patients need to take medication for many months even after symptoms disappear (at least 6 months under direct observation treatment (DOT) [53]. The disease is often fatal if left untreated (about 50 per cent of HIV-uninfected people and almost all HIV-positive people die of TB without proper treatment) [10]. The four front-line anti-TB drugs target M.tb via different mechanisms of action [42]. Briefly, INH is a prodrug that, after activation by katG, a mycobacterial catalase/peroxidase enzyme, inhibits the synthesis of mycolic acids which are essential components of the mycobacterial cell wall [54]. Rifampicin, also known as rifampin, exerts its antimicrobial activity by forming a stable complex with the bacterial DNA-dependent RNA polymerase, thereby inhibiting RNA synthesis [55]. The mechanism of action of PZA is only partially understood, despite its clinical use as an anti-tuberculosis drug for 50 years. Pyrazinamide (PZA) is a drug that is converted to pyrazinoic acid (POA) by the bacterial enzyme pyrazinamidase. POA inhibits the bacterial synthesis of coenzyme A, an essential cofactor in metabolism. PZA exerts its effect by penetrating into all types of TB lung lesions, including necrotic caseous granulomas, and by killing the non-growing bacilli [56]. Ethambutol is a bacteriostatic agent that inhibits the arabinosyl transferases (embA, embB, and embC), which are essential for the synthesis of the bacterial cell wall arabinogalactan and lipoarabinomannan [57]. An in-depth description of the mechanisms of action of four drugs that target M.tb mechanisms of action has been reported elsewhere [42]. Although treatment with all “first-line TB drugs” for 2 months, followed by 4 months of isoniazid and rifampicin, which is used to treat DS-TB, had an overall success rate of 86% in 2019, there are still unresolved problems with the duration of this therapy [42][58]. In fact, despite the gradual reduction in the duration of therapy required to treat DS-TB from 24 to 6 months, non-compliance or dropout remain major barriers to effective treatment. Unfortunately, this depends on the various adverse effects associated with this treatment, including hepatitis, skin reactions, gastrointestinal intolerance, neurological, and hematological toxicity [42].
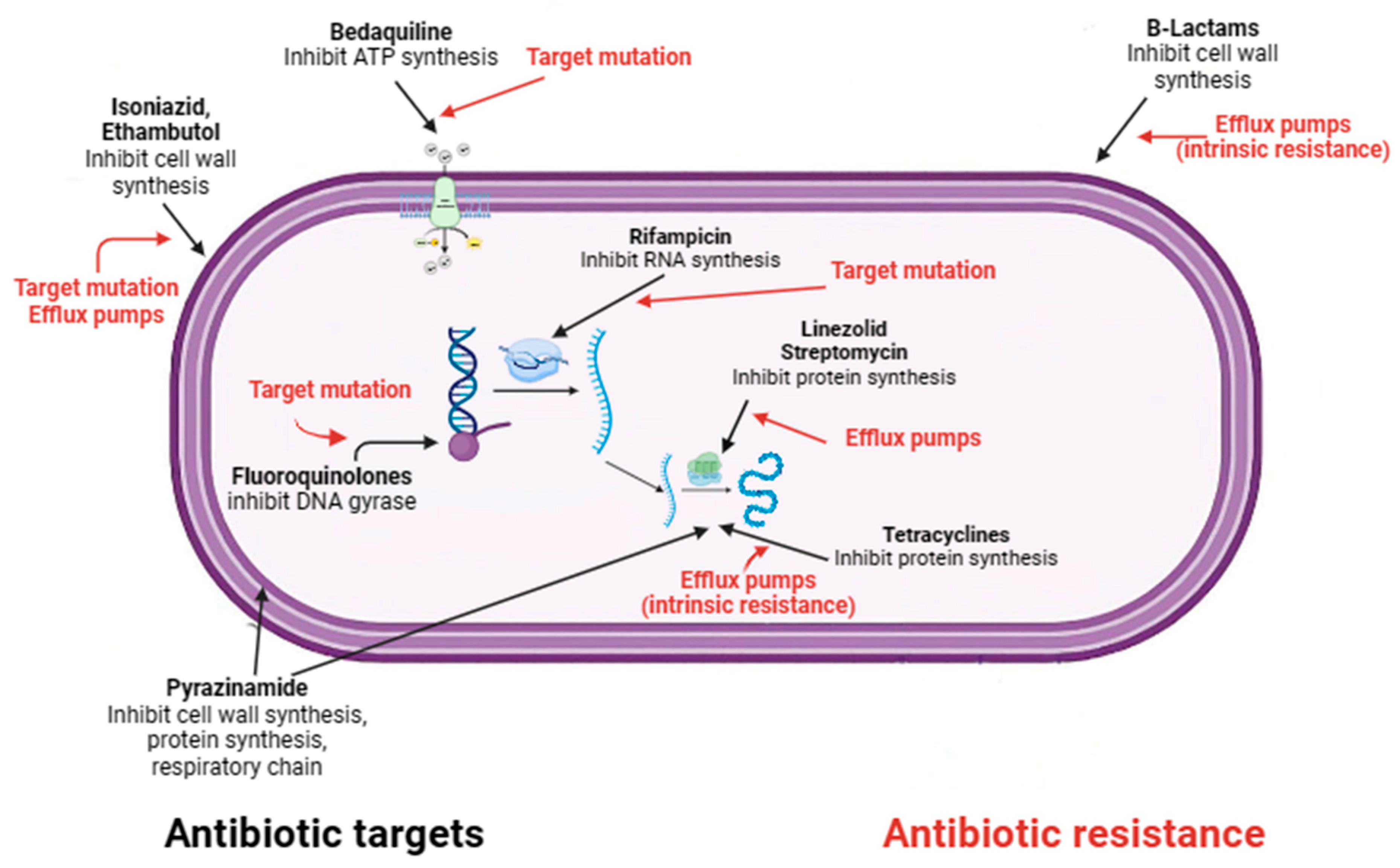
Figure 2. Mechanisms of action and resistance of antibiotics in M. tuberculosis.
This entry is adapted from the peer-reviewed paper 10.3390/microorganisms11092277
References
- Shuaib, Y.A.; Utpatel, C.; Kohl, T.A.; Barilar, I.; Diricks, M.; Ashraf, N.; Wieler, L.H.; Kerubo, G.; Mesfin, E.A.; Diallo, A.B.; et al. Origin and Global Expansion of Mycobacterium tuberculosis Complex Lineage 3. Genes 2022, 13, 990.
- Hargreaves, J.R.; Boccia, D.; Evans, C.A.; Adato, M.; Petticrew, M.; Porter, J.D. The social determinants of tuberculosis: From evidence to action. Am. J. Public Health 2011, 101, 654–662.
- Zwerling, A.; Hanrahan, C.; Dowdy, D.W. Ancient Disease, Modern Epidemiology: A Century of Progress in Understanding and Fighting Tuberculosis. Am. J. Epidemiol. 2016, 183, 407–414.
- Barry, C.E. Lessons from seven decades of antituberculosis drug discovery. Curr. Top. Med. Chem. 2011, 11, 1216–1225.
- Cegielski, J.P. Extensively drug-resistant tuberculosis: “There must be some kind of way out of here”. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2010, 50 (Suppl. 3), S195–S200.
- Yang, Q.; Han, J.; Shen, J.; Peng, X.; Zhou, L.; Yin, X. Diagnosis and treatment of tuberculosis in adults with HIV. Medicine 2022, 101, e30405.
- Glaziou, P.; Floyd, K.; Raviglione, M.C. Global Epidemiology of Tuberculosis. Semin. Respir. Crit. Care Med. 2018, 39, 271–285.
- Chakaya, J.; Petersen, E.; Nantanda, R.; Mungai, B.N.; Migliori, G.B.; Amanullah, F.; Lungu, P.; Ntoumi, F.; Kumarasamy, N.; Maeurer, M.; et al. The WHO Global Tuberculosis 2021 Report—Not so good news and turning the tide back to End TB. Int. J. Infect. Dis. IJID Off. Publ. Int. Soc. Infect. Dis. 2022, 124 (Suppl. 1), S26–S29.
- Fukunaga, R.; Glaziou, P.; Harris, J.B.; Date, A.; Floyd, K.; Kasaeva, T. Epidemiology of Tuberculosis and Progress Toward Meeting Global Targets—Worldwide, 2019. MMWR. Morb. Mortal. Wkly. Rep. 2021, 70, 427–430.
- Hamada, Y.; Getahun, H.; Tadesse, B.T.; Ford, N. HIV-associated tuberculosis. Int. J. STD AIDS 2021, 32, 780–790.
- Cai, L.; Hu, X.; Huang, Y.; Huang, X.; Tong, Y. Editorial: Updates on tuberculosis control and management. Front. Public Health 2022, 10, 1126429.
- Bloom, B.R.; Atun, R.; Cohen, T.; Dye, C.; Fraser, H.; Gomez, G.B.; Knight, G.; Murray, M.; Nardell, E.; Rubin, E.; et al. Tuberculosis. In Major Infectious Diseases, 3rd ed.; Holmes, K.K., Bertozzi, S., Bloom, B.R., Jha, P., Eds.; The International Bank for Reconstruction and Development/The World Bank: Washington, DC, USA, 2017; p. 11.
- Chakaya, J.; Khan, M.; Ntoumi, F.; Aklillu, E.; Fatima, R.; Mwaba, P.; Kapata, N.; Mfinanga, S.; Hasnain, S.E.; Katoto, P.; et al. Global Tuberculosis Report 2020—Reflections on the Global TB burden, treatment and prevention efforts. Int. J. Infect. Dis. IJID Off. Publ. Int. Soc. Infect. Dis. 2021, 113 (Suppl. 1), S7–S12.
- Jang, J.G.; Chung, J.H. Diagnosis and treatment of multidrug-resistant tuberculosis. Yeungnam Univ. J. Med. 2020, 37, 277–285.
- WHO. W.H.O. Tuberculosis; WHO: Geneva, Switzerland, 2023.
- Li, T.; Du, X.; Kang, J.; Luo, D.; Liu, X.; Zhao, Y. Patient, Diagnosis, and Treatment Delays Among Tuberculosis Patients Before and During COVID-19 Epidemic—China, 2018–2022. China CDC Wkly. 2023, 5, 259–265.
- Dheda, K.; Perumal, T.; Moultrie, H.; Perumal, R.; Esmail, A.; Scott, A.J.; Udwadia, Z.; Chang, K.C.; Peter, J.; Pooran, A.; et al. The intersecting pandemics of tuberculosis and COVID-19: Population-level and patient-level impact, clinical presentation, and corrective interventions. Lancet. Respir. Med. 2022, 10, 603–622.
- World Health Organization. Tuberculosis Deaths and Disease Increase during the COVID-19 Pandemic; Global Tuberculosis Report 2022; WHO: Geneva, Switzerland, 2022.
- Dean, A.S.; Tosas Auguet, O.; Glaziou, P.; Zignol, M.; Ismail, N.; Kasaeva, T.; Floyd, K. 25 years of surveillance of drug-resistant tuberculosis: Achievements, challenges, and way forward. Lancet. Infect. Dis. 2022, 22, e191–e196.
- Kostyukova, I.; Pasechnik, O.; Mokrousov, I. Epidemiology and Drug Resistance Patterns of Mycobacterium tuberculosis in High-Burden Area in Western Siberia, Russia. Microorganisms 2023, 11, 425.
- Cummings, K.J. Tuberculosis control: Challenges of an ancient and ongoing epidemic. Public Health Rep. 2007, 122, 683–692.
- Khabibullina, N.F.; Kutuzova, D.M.; Burmistrova, I.A.; Lyadova, I.V. The Biological and Clinical Aspects of a Latent Tuberculosis Infection. Trop. Med. Infect. Dis. 2022, 7, 48.
- Behr, M.A.; Edelstein, P.H.; Ramakrishnan, L. Revisiting the timetable of tuberculosis. Bmj 2018, 362, k2738.
- Pawlowski, A.; Jansson, M.; Skold, M.; Rottenberg, M.E.; Kallenius, G. Tuberculosis and HIV co-infection. PLoS Pathog. 2012, 8, e1002464.
- Patterson, B.; Wood, R. Is cough really necessary for TB transmission? Tuberculosis 2019, 117, 31–35.
- Ryndak, M.B.; Laal, S. Mycobacterium tuberculosis Primary Infection and Dissemination: A Critical Role for Alveolar Epithelial Cells. Front. Cell. Infect. Microbiol. 2019, 9, 299.
- Nardell, E.A. Transmission and Institutional Infection Control of Tuberculosis. Cold Spring Harb. Perspect. Med. 2015, 6, a018192.
- Delogu, G.; Sali, M.; Fadda, G. The biology of mycobacterium tuberculosis infection. Mediterr. J. Hematol. Infect. Dis. 2013, 5, e2013070.
- Chandra, P.; Grigsby, S.J.; Philips, J.A. Immune evasion and provocation by Mycobacterium tuberculosis. Nat. Rev. Microbiol. 2022, 20, 750–766.
- Smith, I. Mycobacterium tuberculosis pathogenesis and molecular determinants of virulence. Clin. Microbiol. Rev. 2003, 16, 463–496.
- Ahmad, F.; Rani, A.; Alam, A.; Zarin, S.; Pandey, S.; Singh, H.; Hasnain, S.E.; Ehtesham, N.Z. Macrophage: A Cell With Many Faces and Functions in Tuberculosis. Front. Immunol. 2022, 13, 747799.
- Cohen, S.B.; Gern, B.H.; Delahaye, J.L.; Adams, K.N.; Plumlee, C.R.; Winkler, J.K.; Sherman, D.R.; Gerner, M.Y.; Urdahl, K.B. Alveolar Macrophages Provide an Early Mycobacterium tuberculosis Niche and Initiate Dissemination. Cell Host Microbe 2018, 24, 439–446.e434.
- Khan, A.; Singh, V.K.; Hunter, R.L.; Jagannath, C. Macrophage heterogeneity and plasticity in tuberculosis. J. Leukoc. Biol. 2019, 106, 275–282.
- Behar, S.M.; Martin, C.J.; Booty, M.G.; Nishimura, T.; Zhao, X.; Gan, H.X.; Divangahi, M.; Remold, H.G. Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis. Mucosal Immunol. 2011, 4, 279–287.
- Martin, C.J.; Booty, M.G.; Rosebrock, T.R.; Nunes-Alves, C.; Desjardins, D.M.; Keren, I.; Fortune, S.M.; Remold, H.G.; Behar, S.M. Efferocytosis is an innate antibacterial mechanism. Cell Host Microbe 2012, 12, 289–300.
- Garcia-Bengoa, M.; Meurer, M.; Goethe, R.; Singh, M.; Reljic, R.; von Kockritz-Blickwede, M. Role of phagocyte extracellular traps during Mycobacterium tuberculosis infections and tuberculosis disease processes. Front. Microbiol. 2023, 14, 983299.
- Ravesloot-Chavez, M.M.; Van Dis, E.; Stanley, S.A. The Innate Immune Response to Mycobacterium tuberculosis Infection. Annu. Rev. Immunol. 2021, 39, 611–637.
- Cooper, A.M. Cell-mediated immune responses in tuberculosis. Annu. Rev. Immunol. 2009, 27, 393–422.
- Esmail, H.; Barry, C.E., 3rd; Wilkinson, R.J. Understanding latent tuberculosis: The key to improved diagnostic and novel treatment strategies. Drug Discov. Today 2012, 17, 514–521.
- Gengenbacher, M.; Kaufmann, S.H. Mycobacterium tuberculosis: Success through dormancy. FEMS Microbiol. Rev. 2012, 36, 514–532.
- Petersen, H.J.; Smith, A.M. The role of the innate immune system in granulomatous disorders. Front. Immunol. 2013, 4, 120.
- Alsayed, S.S.R.; Gunosewoyo, H. Tuberculosis: Pathogenesis, Current Treatment Regimens and New Drug Targets. Int. J. Mol. Sci. 2023, 24, 5202.
- Kim, M.J.; Wainwright, H.C.; Locketz, M.; Bekker, L.G.; Walther, G.B.; Dittrich, C.; Visser, A.; Wang, W.; Hsu, F.F.; Wiehart, U.; et al. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol. Med. 2010, 2, 258–274.
- Ulrichs, T.; Kosmiadi, G.A.; Trusov, V.; Jorg, S.; Pradl, L.; Titukhina, M.; Mishenko, V.; Gushina, N.; Kaufmann, S.H. Human tuberculous granulomas induce peripheral lymphoid follicle-like structures to orchestrate local host defence in the lung. J. Pathol. 2004, 204, 217–228.
- Kanabalan, R.D.; Lee, L.J.; Lee, T.Y.; Chong, P.P.; Hassan, L.; Ismail, R.; Chin, V.K. Human tuberculosis and Mycobacterium tuberculosis complex: A review on genetic diversity, pathogenesis and omics approaches in host biomarkers discovery. Microbiol. Res. 2021, 246, 126674.
- Lee, O.Y.; Wu, H.H.; Donoghue, H.D.; Spigelman, M.; Greenblatt, C.L.; Bull, I.D.; Rothschild, B.M.; Martin, L.D.; Minnikin, D.E.; Besra, G.S. Mycobacterium tuberculosis complex lipid virulence factors preserved in the 17,000-year-old skeleton of an extinct bison, Bison antiquus. PLoS ONE 2012, 7, e41923.
- Forrellad, M.A.; Klepp, L.I.; Gioffre, A.; Sabio y Garcia, J.; Morbidoni, H.R.; de la Paz Santangelo, M.; Cataldi, A.A.; Bigi, F. Virulence factors of the Mycobacterium tuberculosis complex. Virulence 2013, 4, 3–66.
- Kestler, B.; Tyler, S.K. Latent tuberculosis testing through the ages: The search for a sleeping killer. Am. J. Physiol. Lung Cell. Mol. Physiol. 2022, 322, L412–L419.
- Oh, C.E.; Menzies, D. Four months of rifampicin monotherapy for latent tuberculosis infection in children. Clin. Exp. Pediatr. 2022, 65, 214–221.
- Sterling, T.R.; Njie, G.; Zenner, D.; Cohn, D.L.; Reves, R.; Ahmed, A.; Menzies, D.; Horsburgh, C.R., Jr.; Crane, C.M.; Burgos, M.; et al. Guidelines for the Treatment of Latent Tuberculosis Infection: Recommendations from the National Tuberculosis Controllers Association and CDC, 2020. MMWR. Recomm. Rep. Morb. Mortal. Wkly. Rep. Recomm. Rep. 2020, 69, 1–11.
- Assefa, D.G.; Bedru, A.; Zeleke, E.D.; Negash, S.E.; Debela, D.T.; Molla, W.; Mengistu, N.; Woldesenbet, T.T.; Bedane, N.F.; Kajogoo, V.D.; et al. Efficacy and safety of different regimens in the treatment of patients with latent tuberculosis infection: A systematic review and network meta-analysis of randomized controlled trials. Arch. Public Health = Arch. Belg. De Sante Publique 2023, 81, 82.
- WHO Consolidated Guidelines on Tuberculosis: Module 4: Treatment—Drug-Resistant Tuberculosis Treatment, 2022 Update; WHO Guidelines Approved by the Guidelines Review Committee: Geneva, Switzerland, 2022.
- Dartois, V.A.; Rubin, E.J. Anti-tuberculosis treatment strategies and drug development: Challenges and priorities. Nat. Rev.. Microbiol. 2022, 20, 685–701.
- Khan, S.R.; Manialawy, Y.; Siraki, A.G. Isoniazid and host immune system interactions: A proposal for a novel comprehensive mode of action. Br. J. Pharmacol. 2019, 176, 4599–4608.
- Campbell, E.A.; Korzheva, N.; Mustaev, A.; Murakami, K.; Nair, S.; Goldfarb, A.; Darst, S.A. Structural mechanism for rifampicin inhibition of bacterial rna polymerase. Cell 2001, 104, 901–912.
- Gopal, P.; Gruber, G.; Dartois, V.; Dick, T. Pharmacological and Molecular Mechanisms Behind the Sterilizing Activity of Pyrazinamide. Trends Pharmacol. Sci. 2019, 40, 930–940.
- Batt, S.M.; Burke, C.E.; Moorey, A.R.; Besra, G.S. Antibiotics and resistance: The two-sided coin of the mycobacterial cell wall. Cell Surf. 2020, 6, 100044.
- Occhineri, S.; Matucci, T.; Rindi, L.; Tiseo, G.; Falcone, M.; Riccardi, N.; Besozzi, G. Pretomanid for tuberculosis treatment: An update for clinical purposes. Curr. Res. Pharmacol. Drug Discov. 2022, 3, 100128.
This entry is offline, you can click here to edit this entry!