Targeted protein degradation is an attractive technology for cancer treatment due to its ability to overcome the unpredictability of the small molecule inhibitors that cause resistance mutations. Various targeted protein degradation strategies have been developed based on the ubiquitin–proteasome system in the cytoplasm or the autophagy–lysosomal system during endocytosis. Here describe technologies for the targeted inhibition and targeted degradation of the epidermal growth factor receptor (EGFR), one of the major transmembrane proteins responsible for the onset and progression of many types of cancer.
- cancer chemotherapy
- transmembrane receptors
- EGFR
- targeted protein degradation
1. Introduction
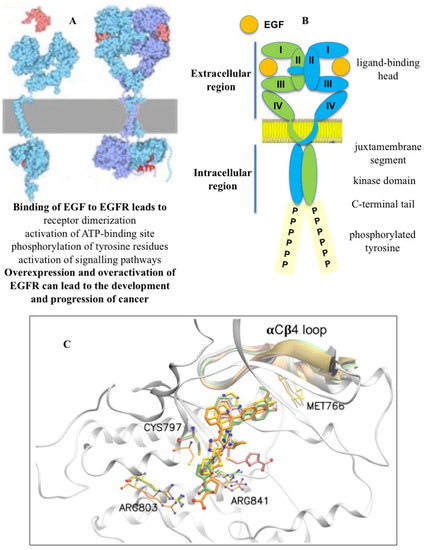
2. Targeted Inhibition of Receptor Tyrosine Kinase EGFR
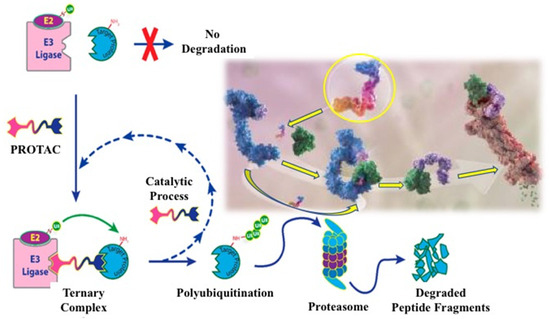
3. EGFR Degradation by PROTAC Technology
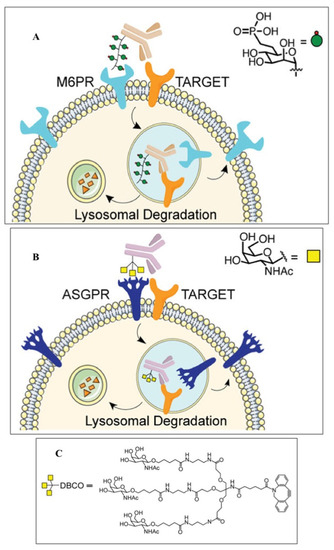
4. Alternative Strategies for EGFR Degradation
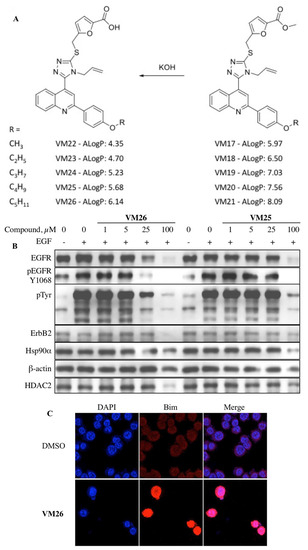
5. New Allosteric Chemicals Bind to EGFR and Lead to Cancer Cell Death
6. New Vision on Cancer Chemotherapy
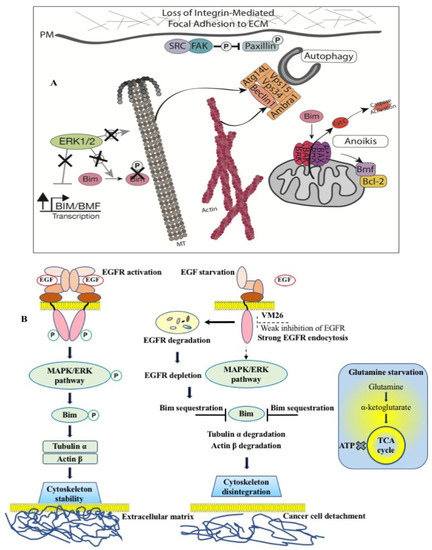
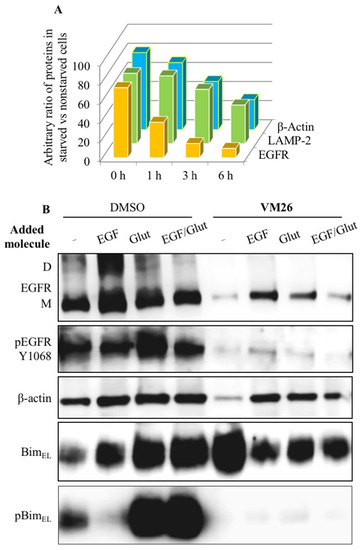
Thus, the data highlight the importance of the alloAUTO strategy in targeting protein degradation through the autophagy–lysosomal and ubiquitin–proteasome pathways. The autophagy response provides the allosteric degradation of the EGFR, leading to a decrease in the activity of the downstream signaling pathways, followed by disabling the phosphorylation of the Bim sensor protein and the destruction of the cellular cytoskeleton, which, ultimately, leads to the death of cancer cells (Figure 6B). The dual mechanism probably reflects the benefits of the cytotoxic effect of standard chemotherapy and the cytostatic effect of targeted chemotherapy on cancer cells, namely, the emergence of fewer resistant mutants in tumor treatment.
This entry is adapted from the peer-reviewed paper 10.3390/biotech12030057
References
- Soneji, S.; Beltrán-Sánchez, H.; Sox, H.C. Assessing progress in reducing the burden of cancer mortality, 1985–2005. J. Clin. Oncol. 2014, 32, 444–448.
- Miller, K.D.; Nogueira, L.; Devasia, T.; Mariotto, A.B.; Yabroff, K.R.; Jemal, A.; Kramer, J.; Siegel, R.L. Cancer treatment and survivorship statistics, 2022. CA Cancer J. Clin. 2022, 72, 409–436.
- Willemsen, M.C.; Mons, U.; Fernández, E. Tobacco control in Europe: Progress and key challenges. Tob. Control 2022, 31, 160–163.
- Patel, V.R.; Qasim Hussaini, S.M.; Blaes, A.H.; Morgans, A.K.; Haynes, A.B.; Adamson, A.S.; Gupta, A. Trends in the prevalence of functional limitations among US cancer survivors, 1999–2018. JAMA Oncol. 2023, 9, 1001–1003.
- Jotte, R.M.; Spigel, D.R. Advances in molecular-based personalized non-small-cell lung cancer therapy: Targeting epidermal growth factor receptor and mechanisms of resistance. Cancer Med. 2015, 4, 1621–1632.
- Castañeda, A.M.; Meléndez, C.M.; Uribe, D.; Pedroza-Díaz, J. Synergistic effects of natural compounds and conventional chemotherapeutic agents: Recent insights for the development of cancer treatment strategies. Heliyon 2022, 8, e09519.
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 15–31.
- Patnaik, S.K.; Chandrasekar, M.J.N.; Nagarjuna, P.; Ramamurthi, D.; Swaroop, A.K. Targeting of ErbB1, ErbB2, and their dual targeting using small molecules and natural peptides: Blocking EGFR cell signaling pathways in cancer: A mini-review. Mini Rev. Med. Chem. 2022, 22, 2831–2846.
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022, 12, 31–46.
- Falzone, L.; Salomone, S.; Libra, M. Evolution of cancer pharmacological treatments at the turn of the third millennium. Front. Pharmacol. 2018, 9, 1300.
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58.
- Levantini, E.; Maroni, G.; Del Re, M.; Tenen, D.G. EGFR signaling pathway as therapeutic target in human cancers. Semin. Cancer Biol. 2022, 85, 253–275.
- Pucci, C.; Martinelli, C.; Ciofani, G. Innovative approaches for cancer treatment: Current perspectives and new challenges. Ecancermedicalscience 2019, 13, 961.
- Zhang, C.; Xu, C.; Gao, X.; Yao, Q. Platinum-based drugs for cancer therapy and anti-tumor strategies. Theranostics 2022, 12, 2115–2132.
- Schlessinger, J. Receptor tyrosine kinases: Legacy of the first two decades. Cold Spring Harb. Perspect. Biol. 2014, 6, a008912.
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20.
- Uribe, M.L.; Marrocco, I.; Yarden, Y. EGFR in Cancer: Signaling Mechanisms, Drugs, and Acquired Resistance. Cancers 2021, 13, 2748.
- Dawson, J.P.; Berger, M.B.; Lin, C.C.; Schlessinger, J.; Lemmon, M.A.; Ferguson, K.M. Epidermal growth factor receptor dimerization and activation require ligand-induced conformational changes in the dimer interface. Mol. Cell. Biol. 2005, 25, 7734–7742.
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272.
- Bae, Y.S.; Kang, S.W.; Seo, M.S.; Baines, I.C.; Tekle, E.; Chock, P.B.; Rhee, S.G. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J. Biol. Chem. 1997, 272, 217–221.
- Paulsen, C.E.; Truong, T.H.; Garcia, F.J.; Homann, A.; Gupta, V.; Leonard, S.E.; Carroll, K.S. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat. Chem. Biol. 2011, 8, 57–64.
- Pan, J.; Carroll, K.S. Chemical biology approaches to study protein cysteine sulfenylation. Biopolymers 2014, 101, 165–172.
- Sakanyan, V.; Angelini, M.; Le Béchec, M.; Lecocq, M.F.; Benaiteau, F.; Rousseau, B.; Gyulkhandanyan, A.; Gyulkhandanyan, L.; Logé, C.; Reiter, E.; et al. Screening and discovery of nitro-benzoxadiazole compounds activating epidermal growth factor receptor (EGFR) in cancer cells. Sci. Rep. 2014, 4, 3977.
- Sakanyan, V.; Hulin, P.; Alves de Sousa, R.; Silva, V.A.; Hambardzumyan, A.; Nedellec, S.; Tomasoni, C.; Logé, C.; Pineau, C.; Roussakis, C.; et al. Activation of EGFR by small compounds through coupling the generation of hydrogen peroxide to stable dimerization of Cu/Zn SOD1. Sci. Rep. 2016, 6, 21088.
- Sakanyan, V.; Benaiteau, F.; Alves de Sousa, R.; Pineau, C.; Artaud, I. Straightforward detection of reactive compound binding to multiple proteins in cancer cells: Towards a better understanding of electrophilic stress. Ann. Clin. Exp. Metabol. 2016, 1, 1006.
- Silva, V.A.; Lafont, F.; Benhelli-Mokrani, H.; Breton, M.L.; Hulin, P.; Chabot, T.; Paris, F.; Sakanyan, V.; Fleury, F. Rapid diminution in the level and activity of DNA-dependent protein kinase in cancer cells by a reactive nitro-benzoxadiazole compound. Int. J. Mol. Sci. 2016, 17, 703.
- Sakanyan, V. Reactive Chemicals and Electrophilic Stress in Cancer: A Minireview. High-Throughput 2018, 7, 12.
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473.
- von Zastrow, M.; Sorkin, A. Mechanisms for Regulating and Organizing Receptor Signaling by Endocytosis. Annu. Rev. Biochem. 2021, 90, 709–737.
- Tomas, A.; Futter, C.E.; Eden, E.R. EGF receptor trafficking: Consequences for signaling and cancer. Trends Cell Biol. 2014, 24, 26–34.
- Caldieri, G.; Malabarba, M.G.; Di Fiore, P.P.; Sigismund, S. EGFR Trafficking in Physiology and Cancer. Prog. Mol. Subcell. Biol. 2018, 57, 235–272.
- Zhou, Y.; Sakurai, H. New trend in ligand-induced EGFR trafficking: A dual-mode clathrin-mediated endocytosis model. J. Proteom. 2022, 255, 104503.
- Mendelsohn, J.; Baselga, J. Epidermal growth factor receptor targeting in cancer. Semin. Oncol. 2006, 33, 369–385.
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib. Nat. Rev. Drug Discov. 2021, 20, 551–569.
- Red Brewer, M.; Yun, C.H.; Lai, D.; Lemmon, M.A.; Eck, M.J.; Pao, W. Mechanism for activation of mutated epidermal growth factor receptors in lung cancer. Proc. Natl. Acad. Sci. USA 2013, 110, e3595-604, Erratum in Proc. Natl. Acad. Sci. USA 2013, 110, 20344.
- Arter, C.; Trask, L.; Ward, S.; Yeoh, S.; Bayliss, R. Structural features of the protein kinase domain and targeted binding by small-molecule inhibitors. J. Biol. Chem. 2022, 298, 102247.
- Herbst, R.S.; Fukuoka, M.; Baselga, J. Gefitinib—A novel targeted approach to treating cancer. Nat. Rev. Cancer 2004, 4, 956–965.
- Cappuzzo, F.; Ciuleanu, T.; Stelmakh, L.; Cicenas, S.; Szczésna, A.; Juhász, E.; Esteban, E.; Molinier, O.; Brugger, W.; Melezínek, I.; et al. SATURN investigators. Erlotinib as maintenance treatment in advanced non-small-cell lung cancer: A multicentre, randomised, placebo-controlled phase 3 study. Lancet Oncol. 2010, 11, 521–529.
- Jänne, P.A. Challenges of detecting EGFR T790M in gefitinib/erlotinib-resistant tumours. Lung Cancer 2008, 60 (Suppl. 2), S3–S9.
- Shah, R.; Lester, J.F. Tyrosine kinase inhibitors for the yreatment of EGFR mutation-positive Non-Small-Cell Lung Cancer: A clash of the generations. Clin. Lung Cancer 2020, 21, e216–e228.
- Hirsch, F.R.; Scagliotti, G.V.; Mulshine, J.L.; Kwon, R.; Curran, W.J.; Wu, Y.-L.; Paz-Ares, L. Lung cancer: Current therapies and new targeted treatments. Lancet 2017, 389, 299–311.
- Li, L.; Luo, S.; Lin, H.; Yang, H.; Chen, H.; Liao, Z.; Lin, W.; Zheng, W.; Xie, X. Correlation between EGFR mutation status and the incidence of brain metastases in patients with non-small cell lung cancer. J. Thorac. Dis. 2017, 9, 2510–2520.
- Russo, A.; Franchina, T.; Ricciardi, G.R.R.; Picone, A.; Ferraro, G.; Zanghì, M.; Toscano, G.; Giordano, A.; Adamo, V. A decade of EGFR inhibition in EGFR-mutated non-small-cell lung cancer (NSCLC): Old successes and future perspectives. Oncotarget 2015, 6, 26814–26825.
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Janne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792.
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73.
- Godin-Heymann, N.; Ulkus, L.; Brannigan, B.W.; McDermott, U.; Lamb, J.; Maheswaran, S.; Settleman, J.; Haber, D.A. The T790M “gatekeeper” mutation in EGFR mediates resistance to low concentrations of an irreversible EGFR inhibitor. Mol. Cancer Ther. 2008, 7, 874–879.
- Yun, C.H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075.
- Yu, H.A.; Riely, G.J. Second-generation epidermal growth factor receptor tyrosine kinase inhibitors in lung cancers. J. Natl. Compr. Cancer Netw. 2013, 11, 161–169.
- Meng, Y.; Yu, B.; Huang, H.; Peng, Y.; Li, E.; Yao, Y.; Song, C.; Yu, W.; Zhu, K.; Wang, K.; et al. Discovery of dosimertinib, a highly potent, selective, and orally efficacious deuterated EGFR targeting clinical candidate for the treatment of Non-Small-Cell Lung Cancer. J. Med. Chem. 2021, 64, 925–937.
- Liu, Q.; Sabnis, Y.; Zhao, Z.; Zhang, T.; Buhrlage, S.J.; Jones, L.H.; Gray, N.S. Developing irreversible inhibitors of the protein kinase cysteinome. Chem. Biol. 2013, 20, 146–159.
- da Cunha Santos, G.; Shepherd, F.A.; Tsao, M.S. EGFR mutations and lung cancer. Annu. Rev. Pathol. 2011, 6, 49–69.
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454.
- Huang, F.; Han, X.; Xiao, X.; Zhou, J. Covalent warheads targeting cysteine residue: The promising approach in drug development. Molecules 2022, 27, 7728.
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074.
- Finlay, M.R.; Anderton, M.; Ashton, S.; Ballard, P.; Bethel, P.A.; Box, M.R.; Bradbury, R.H.; Brown, S.J.; Butterworth, S.; Campbell, A.; et al. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J. Med. Chem. 2014, 57, 8249–8267.
- Shah, M.P.; Neal, J.W. Targeting acquired and intrinsic resistance mechanisms in Epidermal Growth Factor Receptor mutant Non-Small-Cell Lung Cancer. Drugs 2022, 82, 649–662.
- He, J.; Zhou, Z.; Sun, X.; Yang, Z.; Zheng, P.; Xu, S.; Zhu, W. The new opportunities in medicinal chemistry of fourth-generation EGFR inhibitors to overcome C797S mutation. Eur. J. Med. Chem. 2021, 210, 112995.
- Wang, Z.; Yang, J.J.; Huang, J.; Ye, J.Y.; Zhang, X.C.; Tu, H.Y.; Han-Zhang, H.; Wu, Y.L. Lung adenocarcinoma harboring EGFR T790M and in trans C797S responds to combination therapy of first- and third-generation EGFR TKIs and shifts allelic configuration at resistance. J. Thorac. Oncol. 2017, 12, 1723–1727.
- Jia, Y.; Yun, C.H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H.; et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534, 129–132.
- Wang, S.; Song, Y.; Liu, D. EAI045: The fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett. 2017, 385, 51–54.
- To, C.; Jang, J.; Chen, T.; Park, E.; Mushajiang, M.; De Clercq, D.J.H.; Xu, M.; Wang, S.; Cameron, M.D.; Heppner, D.E.; et al. Single and dual targeting of mutant EGFR with an allosteric inhibitor. Cancer Discov. 2019, 9, 926–943.
- Jang, J.; To, C.; De Clercq, D.J.H.; Park, E.; Ponthier, C.M.; Shin, B.H.; Mushajiang, M.; Nowak, R.P.; Fischer, E.S.; Eck, M.J.; et al. Mutant-selective allosteric EGFR degraders are effective against a broad range of drug-resistant mutations. Angew. Chem. Int. Ed. Engl. 2020, 59, 14481–14489.
- Dong, H.; Ye, X.; Zhu, Y.; Shen, H.; Shen, H.; Chen, W.; Ji, M.; Zheng, M.; Wang, K.; Cai, Z.; et al. Discovery of potent and wild-type-sparing fourth-generation EGFR Inhibitors for treatment of osimertinib-resistance NSCLC. J. Med. Chem. 2023, 66, 6849–6868.
- Marasco, M.; Misale, S. Resistance is futile with fourth-generation EGFR inhibitors. Nat. Cancer 2022, 3, 381–383.
- Xu, L.; Xu, B.; Wang, J.; Gao, Y.; He, X.; Xie, T.; Ye, X.Y. Recent advances of novel fourth generation EGFR inhibitors in overcoming C797S mutation of lung cancer therapy. Eur. J. Med. Chem. 2023, 245 Pt 1, 114900.
- Chirnomas, D.; Hornberger, K.R.; Crews, C.M. Protein degraders enter the clinic—A new approach to cancer therapy. Nat. Rev. Clin. Oncol. 2023, 20, 265–278.
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559.
- Sakamoto, K.M.; Kim, K.B.; Verma, R.; Ransick, A.; Stein, B.; Crews, C.M.; Deshaies, R.J. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol. Cell. Proteom. 2003, 2, 1350–1358.
- Bondeson, D.; Mares, A.; Smith, I.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617.
- Dale, B.; Cheng, M.; Park, K.S.; Kaniskan, H.Ü.; Xiong, Y.; Jin, J. Advancing targeted protein degradation for cancer therapy. Nat. Rev. Cancer 2021, 21, 638–654.
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200.
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435.
- Li, X.; Pu, W.; Zheng, Q.; Ai, M.; Chen, S.; Peng, Y. Proteolysis-targeting chimeras (PROTACs) in cancer therapy. Mol. Cancer 2022, 21, 99.
- Yau, R.; Rape, M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 2016, 18, 579–586.
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and function of the 26S proteasome. Annu. Rev. Biochem. 2018, 87, 697–724.
- Wolska-Washer, A.; Smolewski, P. Targeting protein degradation pathways in tumors: Focusing on their role in hematological malignancies. Cancers 2022, 14, 3778.
- Ishida, T.; Ciulli, A. E3 ligase ligands for PROTACs: How they were found and How to discover new ones. SLAS Discov. 2021, 26, 484–502.
- Li, M.; Zhi, Y.; Liu, B.; Yao, Q. Advancing strategies for proteolysis-targeting chimera design. J. Med. Chem. 2023, 66, 2308–2329.
- Smith, B.E.; Wang, S.L.; Jaime-Figueroa, S.; Harbin, A.; Wang, J.; Hamman, B.D.; Crews, C.M. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun. 2019, 10, 131.
- Kofink, C.; Trainor, N.; Mair, B.; Wohrle, S.; Wurm, M.; Mischerikow, N.; Roy, M.J.; Bader, G.; Greb, P.; Garavel, G.; et al. A selective and orally bioavailable VHL-recruiting PROTAC achieves SMARCA2 degradation in vivo. Nat. Commun. 2022, 13, 5969.
- Lu, J.; Huang, Y.; Huang, J.; He, R.; Huang, M.; Lu, X.; Xu, Y.; Zhou, F.; Zhang, Z.; Ding, K. Discovery of the first examples of threonine tyrosine kinase PROTAC degraders. J. Med. Chem. 2022, 65, 2313–2328.
- Hu, J.; Hu, B.; Wang, M.; Xu, F.; Miao, B.; Yang, C.-Y.; Wang, M.; Liu, Z.; Hayes, D.F.; Chinnaswamy, K.; et al. Discovery of ERD-308 as a highly potent proteolysis targeting chimera (PROTAC) degrader of estrogen receptor (ER). J. Med. Chem. 2019, 62, 1420–1442.
- Yokoo, H.; Shibata, N.; Naganuma, M.; Murakami, Y.; Fujii, K.; Ito, T.; Aritake, K.; Naito, M.; Demizu, Y. Development of a hematopoietic prostaglandin D synthase-degradation inducer. ACS Med. Chem. Lett. 2021, 12, 236–241.
- Khan, S.; Zhang, X.; Lv, D.; Zhang, Q.; He, Y.; Zhang, P.; Liu, X.; Thummuri, D.; Yuan, Y.; Wiegand, J.S.; et al. A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat. Med. 2019, 25, 1938–1947.
- Li, X.; Song, Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J. Hematol. Oncol. 2020, 13, 50.
- Zhao, Q.; Lan, T.; Su, S.; Rao, Y. Induction of apoptosis in MDAMB-231 breast cancer cells by a PARP1-targeting PROTAC small molecule. Chem. Commun. 2019, 55, 369–372.
- Yan, W.; Pan, B.; Shao, J.; Lin, H.; Li, H. Feasible column chromatography-free, multi-gram scale synthetic process of VH032 amine, which could enable rapid PROTAC library construction. ACS Omega 2022, 7, 26015–26020.
- Burslem, G.M.; Smith, B.E.; Lai, A.C.; Jaime-Figueroa, S.; McQuaid, D.C.; Bondeson, D.P.; Toure, M.; Dong, H.; Qian, Y.; Wang, J.; et al. The advantages of targeted protein degradation over inhibition: An RTK case study. Cell Chem. Biol. 2018, 25, 67–77.e3.
- Jones, L.H.; Mitchell, C.A.; Loberg, L.; Pavkovic, M.; Rao, M.; Roberts, R.; Stamp, K.; Volak, L.; Wittwer, M.B.; Pettit, S. Targeted protein degraders: A call for collective action to advance safety assessment. Nat. Rev. Drug Discov. 2022, 21, 401–402.
- Zeng, S.; Huang, W.; Zheng, X.; Cheng, L.; Zhang, Z.; Wang, J.; Shen, Z. Proteolysis targeting chimera (PROTAC) in drug discovery paradigm: Recent progress and future challenges. Eur. J. Med. Chem. 2021, 210, 112981.
- Ottis, P.; Crews, C.M. Proteolysis-targeting chimeras: Induced protein degradation as a therapeutic strategy. ACS Chem. Biol. 2017, 12, 892–898.
- Bondeson, D.P.; Smith, B.E.; Burslem, G.M.; Buhimschi, A.D.; Hines, J.; Jaime-Figueroa, S.; Wang, J.; Hamman, B.D.; Ishchenko, A.; Crews, C.M. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem. Biol. 2018, 25, 78–87.e5.
- Cromm, P.M.; Samarasinghe, K.T.G.; Hines, J.; Crews, C.M. Addressing kinase-independent functions of Fak via PROTAC mediated degradation. J. Am. Chem. Soc. 2018, 140, 17019–17026.
- Cheng, M.; Yu, X.; Lu, K.; Xie, L.; Wang, L.; Meng, F.; Han, X.; Chen, X.; Liu, J.; Xiong, Y.; et al. Discovery of potent and selective epidermal growth factor receptor (EGFR) bifunctional small-molecule degraders. J. Med. Chem. 2020, 63, 1216–1232.
- He, K.; Zhang, Z.; Wang, W.; Zheng, X.; Wang, X.; Zhang, X. Discovery and biological evaluation of proteolysis targeting chimeras (PROTACs) as EGFR degraders based on osimertinib and lenalidomide. Bioorg. Med. Chem. Lett. 2020, 30, 127167.
- Qu, X.; Liu, H.; Song, X.; Sun, N.; Zhong, H.; Qiu, X.; Yang, X.; Jiang, B. Effective degradation of EGFRL858R+T790M mutant proteins by CRBN-based PROTACs through both proteosome and autophagy/lysosome degradation systems. Eur. J. Med. Chem. 2021, 218, 113328.
- Zhang, X.; Xu, F.; Tong, L.; Zhang, T.; Xie, H.; Lu, X.; Ren, X.; Ding, K. Design and synthesis of selective degraders of EGFRL858R/T790M mutant. Eur. J. Med. Chem. 2020, 192, 112199.
- Zhao, H.-Y.; Yang, X.-Y.; Lei, H.; Xi, X.-X.; Lu, S.-M.; Zhang, J.-J.; Xin, M.; Zhang, S.-Q. Discovery of potent small molecule PROTACs targeting mutant EGFR. Eur. J. Med. Chem. 2020, 208, 112781.
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728.
- Zhang, H.; Zhao, H.-Y.; Xi, X.-X.; Liu, Y.-J.; Xin, M.; Mao, S.; Zhang, J.-J.; Lu, A.X.; Zhang, S.-Q. Discovery of potent epidermal growth factor receptor (EGFR) degraders by proteolysis targeting chimera (PROTAC). Eur. J. Med. Chem. 2020, 189, 112061.
- Zhao, H.-Y.; Wang, H.P.; Mao, Y.Z.; Zhang, H.; Xin, M.; Xi, X.-X.; Lei, H.; Mao, S.; Li, D.H.; Zhang, S.-Q. Discovery of potent PROTACs targeting EGFR mutants through the optimization of covalent EGFR ligands. J. Med. Chem. 2022, 65, 4709–4726.
- Ricordel, C.; Friboulet, L.; Facchinetti, F.; Soria, J.C. Molecular mechanisms of acquired resistance to third-generation EGFR-TKIs in EGFR T790M-mutant lung cancer. Ann. Oncol. 2018, 29 (Suppl. 1), 128–137.
- Wang, S.H.; Tsui, S.T.; Liu, C.; Song, Y.P.; Liu, D.L. EGFR C797S mutation mediates resistance to third-generation inhibitors in T790M-positive non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 59.
- Zhang, H.; Xie, R.; AI-Furas, H.; Li, Y.; Wu, Q.; Li, J.; Xu, F.; Xu, T. Design, synthesis, and biological evaluation of novel EGFR PROTACs targeting del19/T790M/C797S mutation. ACS Med. Chem. Lett. 2022, 13, 278–283.
- Chen, Y.; Tandon, I.; Heelan, W.; Wang, Y.X.; Tang, W.P.; Hu, Q.Y. Proteolysis-targeting chimera (PROTAC) delivery system: Advancing protein degraders towards clinical translation. Chem. Soc. Rev. 2022, 51, 5330–5350.
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.K.; Wang, J.; Chen, X.; Dong, H.Q.; Siu, K.; et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129.
- Moreau, K.; Coen, M.; Zhang, A.X.; Pachl, F.; Castaldi, M.P.; Dahl, G.; Boyd, H.; Scott, C.; Newham, P. Proteolysis-targeting chimeras in drug development: A safety perspective. Brit. J. Pharmacol. 2020, 177, 1709–1718.
- Hong, K.B.; An, H. Degrader-antibody conjugates: Emerging new modality. J. Med. Chem. 2023, 66, 140–148.
- Li, K.; Crews, C.M. PROTACs: Past, present and future. Chem. Soc. Rev. 2022, 51, 5214–5236.
- Dragovich, P.S. Degrader-antibody conjugates. Chem. Soc. Rev. 2022, 51, 3886–3897.
- Yu, N.; Huang, L.; Zhou, Y.; Xue, T.; Chen, Z.; Han, G. Near-infrared-light activatable nanoparticles for deep-tissue-penetrating wireless optogenetics. Adv. Healthc. Mater. 2019, 8, e1801132.
- Zhang, Y.; Zhang, Y.; Song, G.; He, Y.; Zhang, X.; Liu, Y.; Ju, H. A DNA-azobenzene nanopump fueled by upconversion luminescence for controllable intracellular drug release. Angew. Chem. Int. Ed. Engl. 2019, 58, 18207–18211.
- He, Q.; Zhou, L.; Yu, D.; Zhu, R.; Chen, Y.; Song, M.; Liu, X.; Liao, Y.; Ding, T.; Fan, W.; et al. Near-infrared-activatable PROTAC nanocages for controllable target protein degradation and on-demand antitumor therapy. J. Med. Chem. 2023, 66, 10458–10472.
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int. J. Oncol. 2019, 54, 407–419.
- Pirker, R. EGFR-directed monoclonal antibodies in non-small cell lung cancer. Target. Oncol. 2013, 8, 47–53.
- Tabasinezhad, M.; Omidinia, E.; Talebkhan, Y.; Omrani, M.D.; Mahboudi, F.; Ghaedi, H.; Wenzel, W. The effects of somatic mutations on EGFR interaction with anti-EGFR monoclonal antibodies: Implication for acquired resistance. Proteins 2020, 88, 3–14.
- Capdevila, J.; Elez, E.; Macarulla, T.; Ramos, F.J.; Ruiz-Echarri, M.; Tabernero, J. Anti-epidermal growth factor receptor monoclonal antibodies in cancer treatment. Cancer Treat. Rev. 2009, 35, 354–363.
- Ermondi, G.; Garcia-Jimenez, D.; Caron, G. PROTACs and building blocks: The 2D chemical space in very early drug discovery. Molecules 2021, 26, 672.
- Weng, G.; Cai, X.; Cao, D.; Du, H.; Shen, C.; Deng, Y.; He, Q.; Yang, B.; Li, D.; Hou, T. PROTAC-DB 2.0: An updated database of PROTACs. Nucleic Acids Res. 2023, 51, D1367–D1372.
- O’Brien Laramy, M.N.; Luthra, S.; Brown, M.F.; Bartlett, D.W. Delivering on the promise of protein degraders. Nat. Rev. Drug Discov. 2023, 22, 410–427.
- Li, S.; de Camargo Correia, G.S.; Wang, J.; Manochakian, R.; Zhao, Y.; Lou, Y. Emerging targeted therapies in advanced Non-Small-Cell Lung Cancer. Cancers 2023, 15, 2899.
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome- targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584, 291–297.
- Li, Z.; Zhu, C.; Ding, Y.; Fei, Y.; Lu, B. ATTEC: A potential new approach to target proteinopathies. Autophagy 2020, 16, 185–187.
- Takahashi, D.; Arimoto, H. Targeting selective autophagy by AUTAC degraders. Autophagy 2020, 16, 765–766.
- Ji, C.H.; Lee, M.J.; Kim, H.Y.; Heo, A.J.; Park, D.Y.; Kim, Y.K.; Kim, B.Y.; Kwon, Y.T. Targeted protein degradation via the autophagy-lysosome system: AUTOTAC (AUTOphagy-TArgeting Chimera). Autophagy 2022, 18, 2259–2262.
- Itoh, Y.; Ishikawa, M.; Naito, M.; Hashimoto, Y. Protein knockdown using methyl bestatin-ligand hybrid molecules: Design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J. Am. Chem. Soc. 2010, 132, 5820–5826.
- Ahn, G.; Banik, S.M.; Bertozzi, C.R. Degradation from the outside in: Targeting extracellular and membrane proteins for degradation through the endolysosomal pathway. Cell Chem. Biol. 2021, 28, 1072–1080.
- Hong, D.; Zhou, B.; Zhang, B.; Ren, H.; Zhu, L.; Zheng, G.; Ge, M.; Ge, J. Recent advances in the development of EGFR degraders: PROTACs and LYTACs. Eur. J. Med. Chem. 2022, 239, 114533.
- Maity, P.; Chatterjee, J.; Patil, K.T.; Arora, S.; Katiyar, M.K.; Kumar, M.; Samarbakhsh, A.; Joshi, G.; Bhutani, P.; Chugh, M.; et al. Targeting the epidermal growth factor receptor with molecular degraders: State-of-the-art and future opportunities. J. Med. Chem. 2023, 66, 3135–3172.
- Li, X.; Liu, Q.; Xie, X.; Peng, C.; Pang, Q.; Liu, B.; Han, B. Application of novel degraders employing autophagy for expediting medicinal research. J. Med. Chem. 2023, 66, 1700–1711.
- Ahn, G.; Banik, S.M.; Miller, C.L.; Riley, N.M.; Cochran, J.R.; Bertozzi, C.R. LYTACs that engage the asialoglycoprotein receptor for targeted protein degradation. Nat. Chem. Biol. 2021, 17, 937–946.
- Iradyan, M.; Iradyan, N.; Hulin, P.; Hambardzumyan, A.; Gyulkhandanyan, A.; Alves de Sousa, R.; Hessani, A.; Roussakis, C.; Bollot, G.; Bauvais, C.; et al. Targeting degradation of EGFR through the allosteric site leads to cancer cell detachment-promoted death. Cancers 2019, 11, 1094.
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948.
- Tanida, I.; Ueno, T.; Kominami, E. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell. Biol. 2004, 36, 2503–2518.
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528.
- Miyata, Y.; Nakamoto, H.; Neckers, L. The therapeutic target Hsp90 and cancer hallmarks. Curr. Pharm. Des. 2013, 19, 347–365.
- Citri, A.; Harari, D.; Shohat, G.; Ramakrishnan, P.; Gan, J.; Lavi, S.; Eisenstein, M.; Kimchi, A.; Wallach, D.; Pietrokovski, S.; et al. Hsp90 recognizes a common surface on client kinases. J. Biol. Chem. 2006, 281, 14361–14369.
- Pick, E.; Kluger, Y.; Giltnane, J.M.; Moeder, C.; Camp, R.L.; Rimm, D.L.; Kluger, H.M. High HSP90 expression is associated with decreased survival in breast cancer. Cancer Res. 2007, 67, 2932–2937.
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta 2013, 1833, 3481–3498.
- Vlahakis, A.; Debnath, J. The Interconnections between autophagy and integrin-mediated cell adhesion. J. Mol. Biol. 2017, 429, 515–530.
- Reginato, M.J.; Mills, K.R.; Paulus, J.K.; Lynch, D.K.; Sgroi, D.C.; Debnath, J.; Muthuswamy, S.K.; Brugge, J.S. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat. Cell Biol. 2003, 5, 733–740.
- Chi, X.; Nguyen, D.; Pemberton, J.M.; Osterlund, E.J.; Liu, Q.; Brahmbhatt, H.; Zhang, Z.; Lin, J.; Leber, B.; Andrews, D.W. The carboxyl-terminal sequence of bim enables bax activation and killing of unprimed cells. eLife 2020, 9, e44525.
- O’Connor, L.; Strasser, A.; O’Reilly, L.A.; Hausmann, G.; Adams, J.M.; Cory, S.; Huang, D.C. Bim: A novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 1998, 17, 384–395.
- Wilfling, F.; Weber, A.; Potthoff, S.; Vögtle, F.N.; Meisinger, C.; Paschen, S.A.; Häcker, G. BH3-only proteins are tail-anchored in the outer mitochondrial membrane and can initiate the activation of bax. Cell Death Differ. 2012, 19, 1328–1336.
- Gogada, R.; Yadav, N.; Liu, J.; Tang, S.; Zhang, D.; Schneider, A.; Seshadri, A.; Sun, L.; Aldaz, C.M.; Tang, D.G.; et al. Bim, a proapoptotic protein, up-regulated via transcription factor E2F1-dependent mechanism, functions as a prosurvival molecule in cancer. J. Biol. Chem. 2013, 288, 368–381.
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193.
- Tong, J.; Taylor, P.; Moran, M.F. Proteomic analysis of the epidermal growth factor receptor (EGFR) interactome and post-translational modifications associated with receptor endocytosis in response to EGF and stress. Mol. Cell. Proteom. 2014, 13, 1644–1658.
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541.
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364.
- Yan, G.; Elbadawi, M.; Efferth, T. Multiple cell death modalities and their key features (Review). World Acad. Sci. J. 2020, 2, 39–48.
- Le Gall, M.; Chambard, J.C.; Breittmayer, J.P.; Grall, D.; Pouyssegur, J.; Obberghen-Schilling, E. The p42/p44 MAP kinase pathway prevents apoptosis induced by anchorage and serum removal. Mol. Biol. Cell 2000, 11, 1103–1112.
- Quadros, M.R.; Peruzzi, F.; Kari, C.; Rodeck, U. Complex regulation of signal transducers and activators of transcription 3 activation in normal and malignant keratinocytes. Cancer Res. 2004, 64, 3934–3939.
- Chamberlain, P.P.; Hamann, L.G. Development of targeted protein degradation therapeutics. Nat. Chem. Biol. 2019, 15, 937–944.
- Iradyan, M.A.; Iradyan, N.S.; Hambardzumyn, A.A.; Panosyan, H.A.; Tamazyan, R.A.; Ayvazyan, A.G.; Hovhannisyan, G.S.; Alves de sousa, R.; Sakanyan, V.A. Selective N-, S-alkylation of 4-allyl-3--4,5-dihydro-1h-1,2,4-triazole-5-thiones with substituted benzylchlorides. synthesis, docking analysis and cytotoxic action. Chem. J. Arm. 2018, 71, 389–406.
- Iradyan, M.A.; Iradyan, N.S.; Hambardzumyan, A.A.; Minasyan, N.S.; Roussakis, C.; Sakanyan, V.A. Synthesis of furfuryl derivatives of 4-allyl-1-(4-hydroxy-3-nitrobenzyl)-3--4,5-dihydro-1H-1,2,4-triazole-5-thions and their toxicity in cancer cells. Chem. J. Arm. 2018, 71, 559–570.
- Iradyan, M.A.; Iradyan, N.S.; Hambardzumyan, A.A.; Nersesyan, L.E.; Aharonyan, A.S.; Danielyan, I.S.; Muradyan, R.E.; Paronikyan, R.V.; Stepanyan, G.M. Docking analysis and some biological properties of furfuryl derivatives of 4-allyl-5--4H-1,2,4-triazol-3-thiol. Biol. J. Arm. 2018, 70, 100–107.
- Sakanyan, V.A.; Iradyan, M.A.; Iradyan, N.S. Development of targeted EGFR degradation for cancer treatment. Nat. Acad. Sci. Armen. Rep. 2022, 122, 218–227.