The chemical nature of intracellular labile iron pools (LIPs) is described. By virtue of the kinetic lability of these pools, it is suggested that the isolation of such species by chromatography methods will not be possible, but rather mass spectrometric techniques should be adopted. Iron-sensitive fluorescent probes, which have been developed for the detection and quantification of LIP, are described, including those specifically designed to monitor cytosolic, mitochondrial, and lysosomal LIPs. The potential of near-infrared (NIR) probes for in vivo monitoring of LIP is discussed.
- labile iron pools
- fluorescence
- iron sensitive optical probes
- near-infrared probes
- imaging
1. Introduction
2. Chemical Components of LIP
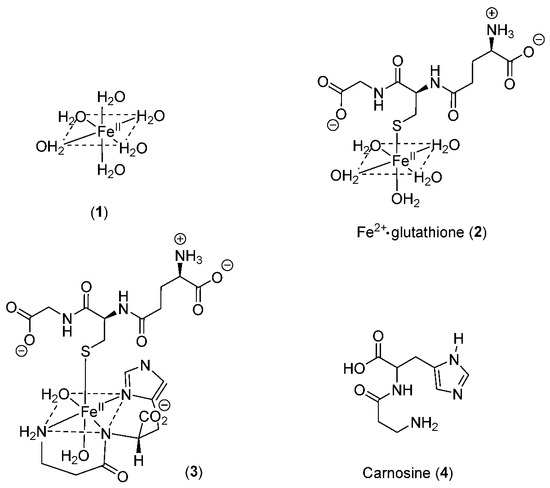
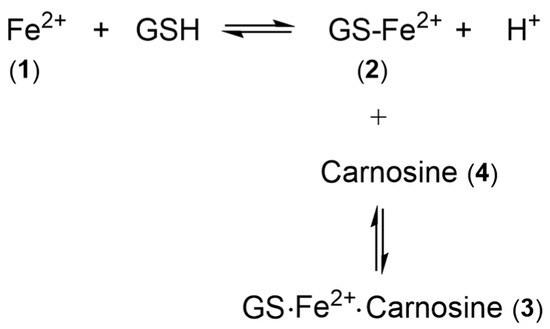
3. The Nature of the Labile Iron Pool in Different Intracellular Organelles
4. Isolation and Characterization of Labile Iron Species
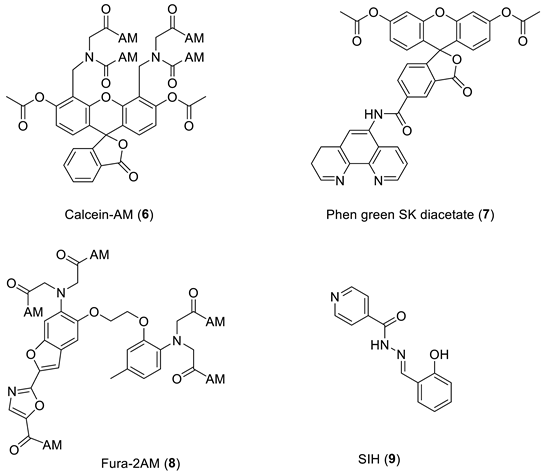
5. Iron-Sensitive Fluorescent Probes

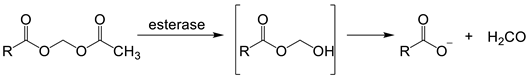
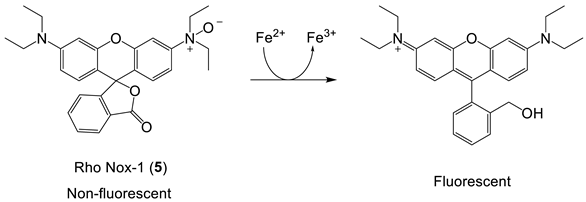
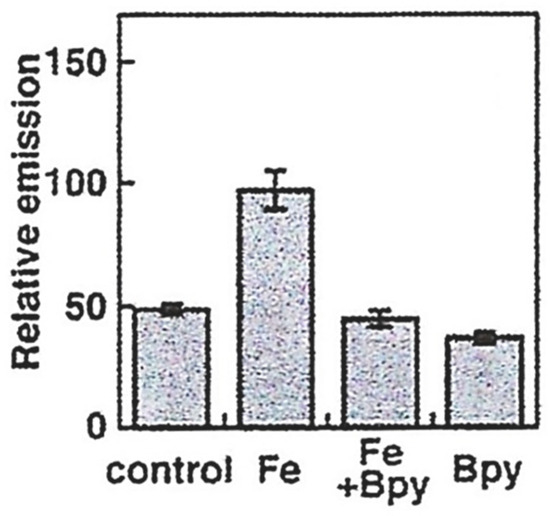
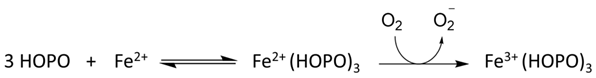
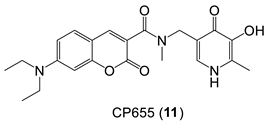
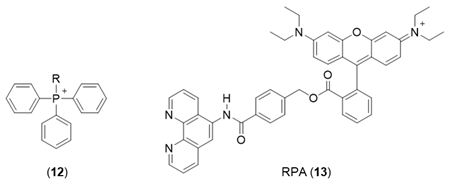
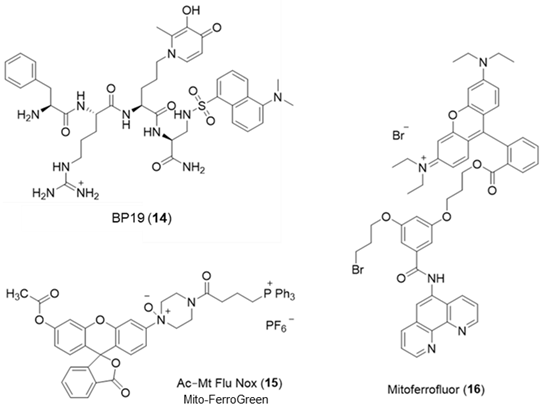
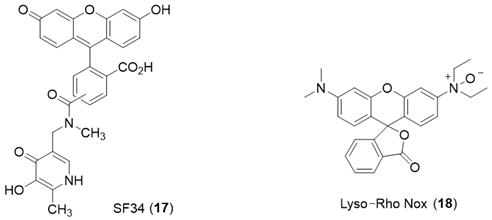
6. NIR Probes
7. Conclusions
The potential of using iron-sensitive probes for the quantification of LIP is considered, and a range of applications of such probes is described. The quantification of LIP by the use of iron-specific fluorescent probes is currently under development, but it is now possible to monitor LIP levels in cytosolic, mitochondrial, and lysosomal pools of cells in suspension. Such measurements made under in vivo conditions will be more difficult, but NIR probes are under development for this purpose. Undoubtedly, the ability to visualize and quantify iron dynamics in living organisms is expected to have a significant impact on the development of targeted therapeutic interventions for iron-related pathological conditions.
This entry is adapted from the peer-reviewed paper 10.3390/molecules28186467
References
- Maret, W.; Wedd, A. Binding, Transport and Storage of Metal Ions in Biological Cells; Royal Society of Chemistry: Cambridge, UK, 2014.
- Hider, R.C.; Kong, X. Iron speciation in the cytosol: An overview. Dalton Trans. 2013, 42, 3220–3229.
- Williams, R.J.P. Free manganese(II) and iron(II) cations can act as intracellular cell controls. FEBS Lett. 1982, 140, 3–10.
- Breuer, W.; Epsztejn, S.; Cabantchik, Z.I. Iron acquired from transferrin by K562 cells is delivered into a cytoplasmic pool of chelatable iron(II). J. Biol. Chem. 1995, 270, 24209–24215.
- Petrat, F.; de Groot, H.; Sustmann, R.; Rauen, U. The chelatable iron pool in living cells: A methodically defined quantity. Biol. Chem. 2002, 383, 489–502.
- Helm, L.; Merbach, A.E. Inorganic and bioinorganic solvent exchange mechanisms. Chem. Rev. 2005, 105, 1922–1959.
- Koppenol, W.H. The centennial of the Fenton reaction. Free Rad. Biol. Med. 1993, 15, 645–651.
- Ran, Y.; Moursy, M.; Hider, R.C.; Cilibrizzi, A. The colorimetric detection of the hydroxyl radical. Int. J. Mol. Sci. 2023, 24, 4162.
- Kühn, L. Iron regulatory proteins and their role in controlling iron metabolism. Metallomics 2015, 7, 232–243.
- Hentze, M.W.; Kühn, L.C. Molecular control of vertebrate iron metabolism: In RNA based circuits operated by iron, nitric oxide and oxidative stress. Proc. Natl. Acad. Sci. USA 1996, 93, 8175–8182.
- Miller, J.P.G.; Perkins, D.J. Model experiments for the study of iron transfer from transferrin to ferritin. Eur. J. Biochem. 1969, 10, 146–151.
- Weaver, J.; Pollack, S. Low-Mr iron isolated from guinea pig reticulocytes as AMP-Fe and ATP-Fe complexes. Biochem. J. 1989, 261, 787–792.
- Veiga, N.; Torres, J.; Mansell, D.; Freeman, S.; Dominguez, S.; Barker, C.J.; Diaz, A.; Kremer, C. “Chelatable iron pool”: Inositol 1,2,3-trisphosphate fulfils the conditions required to be a safe cellular iron ligand. J. Biol. Inorg. Chem. 2009, 14, 51–59.
- Morley, C.G.D.; Bezkorovainy, A. Identification of the iron chelate in hepatocyte cytosol. IRCS Med. Sci. 1983, 11, 1106–1107.
- Hider, R.C.; Kong, X.L. Glutathione: A key component of the cytoplasmic labile iron pool. Biometals 2011, 24, 1179–1187.
- Hamed, M.Y.; Silver, J.; Wilson, M.T. Studies of the reactions of ferric iron with glutathione and some related thiols. Inorg. Chim. Acta 1983, 78, 1–11.
- Fuhr, J.; Rabenstein, D. Nuclear magnetic resonance studies of the solution chemistry of metal complexes. IX binding of cadmium, zinc, lead, and mercury by glutathione. J. Am. Chem. Soc. 1973, 95, 6944–6948.
- Baran, E.J. Metal complexes of carnosine. Biochemistry 2000, 65, 789–797.
- Babitt, J.L.; Huang, F.W.; Xia, Y.; Sidis, Y.; Andrews, N.C.; Lin, H.Y. Modulation of bone morphogenic protein signaling in vivo regulates systemic iron balance. J. Clin. Investig. 2007, 117, 1933–1939.
- Yu, P.B.; Hong, C.C.; Sachidanandan, C.; Babitt, J.L.; Deng, D.Y.; Hoyng, S.A.; Lin, H.Y.; Bloch, K.D.; Peterson, R.T. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat. Chem. Biol. 2008, 4, 33–41.
- Puig, S.; Ramos-Alonso, L.; Romero, A.M.; Martinez-Pastor, M.T. The elemental role of iron in DNA synthesis and repair. Metallomics 2017, 9, 1483–1500.
- O’Keeffe, R.; Latunde-Dada, G.O.; Chen, Y.-L.; Kong, X.L.; Cilibrizzi, A.; Hider, R.C. Glutathione and the intracellular labile heme pool. Biometals 2021, 34, 221–228.
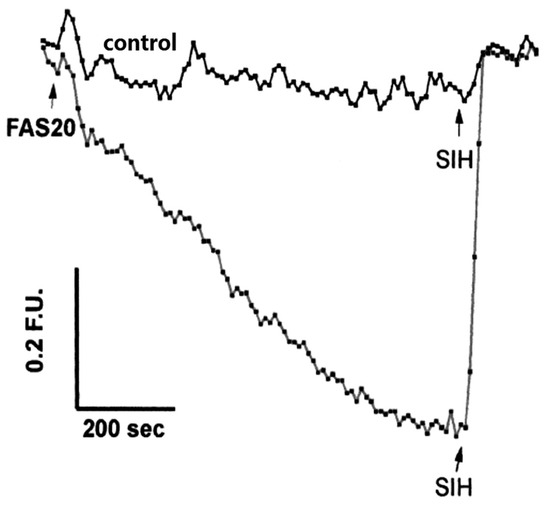
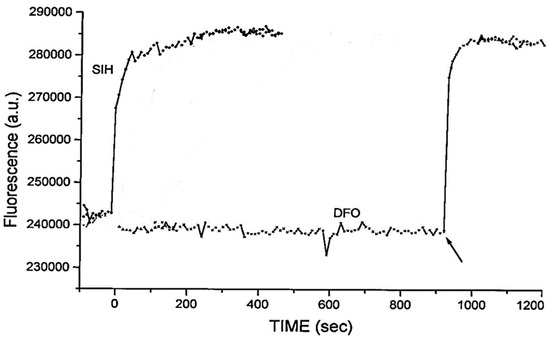
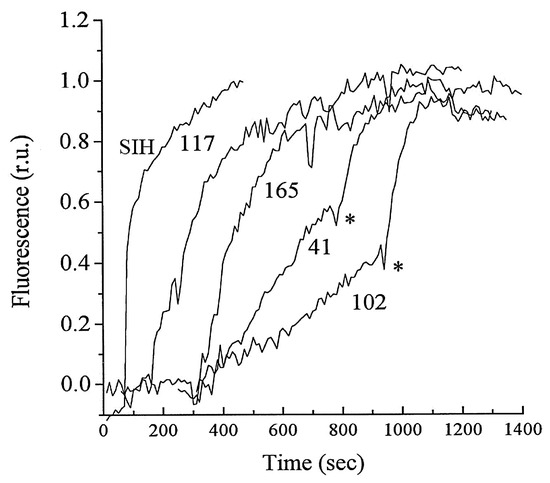
- Ma, Y.; Abbate, V.; Hider, R.C. Iron-sensitive fluorescent probes: Monitoring intracellular iron pools. Metallomics 2015, 7, 212–222.
- Cilibrizzi, A.; Abbate, V.; Chen, Y.-L.; Ma, Y.; Zhou, T.; Hider, R.C. Hydroxypyridinone journey into metal chelation. Chem. Rev. 2018, 118, 7657–7701.
- Maret, W. Analysing free zinc(II) ion concentrations in cell biology with fluorescent chelating molecules. Metallomics 2015, 7, 202–211.
- Alhawsah, B.; Yan, B.; Aydin, Z.; Niu, X.; Guo, M. Highly selective fluorescent probe with an ideal pH profile for the rapid and unambiguous determination of subcellular labile iron (III) pools in human cells. Anal. Lett. 2022, 55, 1954–1970.
- Hirayama, T.; Okuda, K.; Nagasawa, H. A highly selective turn-on fluorescent probe for iron(II) to visualize labile iron in living cells. Chem. Sci. 2013, 4, 1250–1256.
- Hirayama, T.; Tsuboi, H.; Niwa, M.; Miki, A.; Kadota, S.; Ikeshita, Y.; Okua, K.; Nagasawa, H. A universal fluorogenic switch for Fe(II) ion based on N-oxide chemistry permits the visualization of intracellular redox equilibrium shift towards labile iron in hypoxic tumor cells. Chem. Sci. 2017, 8, 4858–4866.
- Carter, K.P.; Young, A.M.; Palmer, A.E. Fluorescent sensors for measuring metal ions in living systems. Chem. Rev. 2014, 114, 4564–4601.
- Hirayama, T.; Nagasawa, H. Chemical tools for detecting Fe ions. J. Clin. Biochem. Nutr. 2017, 60, 39–48.
- Cabantchik, I.; Glickstein, H.; Milgram, P.; Breuer, W. A fluorescence assay for assessing chelation of intracellular iron in a membrane model system and in mammalian cells. Anal. Biochem. 1996, 233, 221–227.
- Glickstein, H.; El, R.B.; Shvartsman, M.; Cabantchik, I. Intracellular labile iron pools as direct targets of iron chelators: A fluorescence study of chelator action in living cells. Blood 2005, 106, 3242–3250.
- Epsztejn, S.; Kakhlon, O.; Glickstein, H.; Breuer, W.; Cabantchik, Z.I. Fluorescence analysis of the labile iron pool of mammalian cells. Anal. Biochem. 1997, 248, 31–40.
- Zanninelli, G.; Glickstein, H.; Breuer, W.; Milgram, P.; Brissot, P.; Hider, R.C.; Konijn, A.M.; Lipman, J.; Shanzer, A.; Cabantchik, Z.I. Chelation and Mobilization of Cellular Iron by Different Classes of Chelators. Mol. Pharmacol. 1997, 51, 842–852.
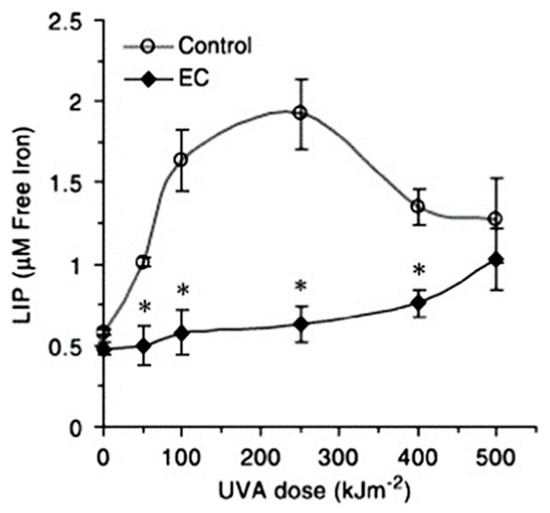
- Basu-Modak, S.; Ali, D.; Gordon, M.; Polte, T.; Yiakouvaki, A.; Pourzand, C.; Rice-Evans, C.; Tyrrell, R.M. Suppression of UVA-mediated release of labile iron by epicatechin—A link to lysosomal protection. Free Radical Biol. Med. 2006, 41, 1197–1204.
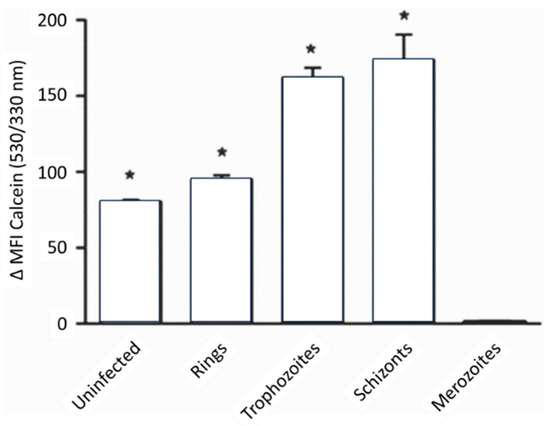
- Clark, M.; Fisher, N.C.; Kasthuri, R.; Hand, C.C. Parasite maturation and host serum iron influence the labile iron pool of erythrocyte stage Plasmodium falciparum. Br. J. Haematol. 2013, 161, 262–269.
- Petrat, F.; Rauen, U.; de Groot, H. Determination of the chelatable iron pool of isolated rat hepatocytes by digital fluorescence microscopy using the fluorescent probe, phen green SK. Hepatology 1999, 29, 1171–1179.
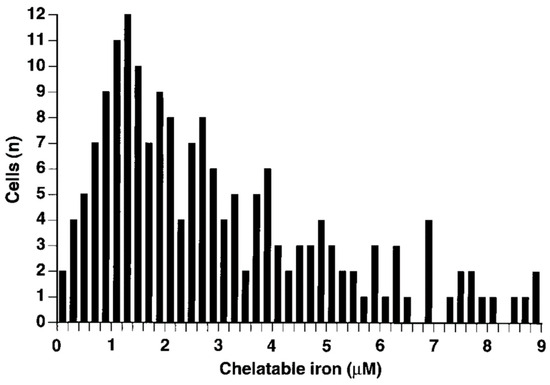
- Petrat, F.; de Groot, H.; Rauen, U. Determination of the chelatable iron pool of single intact cells by laser scanning microscopy. Arch. Biochem. Biophys. 2000, 376, 74–81.
- Monfort, B.; Want, K.; Gervason, S.; D’Autréaux, B. Recent advances in the elucidation of Frataxin biochemical function open novel perspectives for the Treatment of Friedreich’s Ataxia. Front. Neurosci. 2022, 16, 838335.
- Petrat, F.; Weisheit, D.; Lensen, M.; de Groot, H.; Sustmann, R.; Rauen, U. Selective determination of mitochondrial chelatable iron in viable cells with a new fluorescent sensor. Biochem. J. 2002, 362, 137–147.
- Shvartsman, M.; Fibach, E.; Cabantchik, Z.I. Transferrin-iron routing to the cytosol and mitochondria as studied by live and real-time fluorescence. Biochem. J. 2010, 429, 185–193.
- Cabantchik, Z.I. Labile iron in cells and body fluids: Physiology, pathology, and pharmacology. Front. Pharmacol. 2014, 5, 45.
- Reelfs, O.; Abbate, V.; Cilibrizzi, A.; Pook, M.A.; Hider, R.C.; Pourzand, C. The role of mitochondrial labile iron in Friedreich’s ataxia skin fibroblasts sensitivity to ultraviolet A. Metallomics 2019, 11, 656–665.
- Rouault, T.A. Mitochondrial iron overload: Causes and consequences. Curr. Opin. Genet. Dev. 2016, 38, 31–37.
- Kurz, T.; Terman, A.; Gustafsson, B.; Brunk, U.T. Lysosomes and oxidative stress in aging and apoptosis. Biochim. Biophys. Acta 2008, 1780, 1291–1303.
- Kidane, T.Z.; Sauble, E.; Linder, M.C. Release of iron from ferritin requires lysosomal activity. Am. J. Physiol. Cell Physiol. 2006, 291, C445–C455.
- Kürz, T.; Gustafsson, B.; Brunk, U.T. Intralysosomal iron chelation protects against oxidative stress-induced cellular damage. FEBS J. 2006, 273, 3106–3117.
- .Kurz, T.; Terman, A.; Gustafsson, B.; Brunk, U.T. Lysosomes in iron metabolism, ageing and apoptosis. Histochem. Cell. Biol. 2008, 129, 389–406.
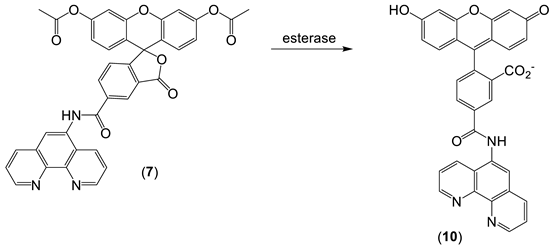
- Fakih, S.; Podinovskaia, M.; Kong, X.; Collins, H.L.; Schaible, U.E.; Hider, R.C. Targeting the Lysosome: Fluorescent Iron(III) Chelators To Selectively Monitor Endosomal/Lysosomal Labile Iron Pools. J. Med. Chem. 2008, 51, 4539–4552.
- Hirayama, T.; Miki, A.; Nagasawa, H. Organelle-specific analysis of labile Fe(II) during ferroptosis by using a cocktail of various colour organelle-targeted fluorescent probes. Metallomics 2019, 11, 111–117.
- Zheng, J.R.; Feng, S.M.; Gong, S.Y.; Xia, Q.F.; Feng, G.Q. In vivo imaging of Fe2+ using an easily obtained probe with a large stokes shift and bright strong lipid droplet-targetable near-infrared fluorescence. Sens. Actuators B Chem. 2020, 309, 127796.
- Dong, B.; Song, W.; Lu, Y.; Tian, M.; Kong, X.; Mehmood, A.H.; Lin, W. Live cell-specific fluorescent probe for the detection of labile Fe(II) and the evaluation of esterase activity in live animals. Sens. Actuators B Chem. 2020, 305, 127470.