Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Interleukin (IL)-23 is a central pro-inflammatory cytokine with a broad range of effects on immune responses. IL-23 is pathologically linked to the induction of the production of the pro-inflammatory cytokines IL-17 and IL-22, which stimulate the differentiation and proliferation of T helper type 17 (Th17) cells. Discoveries suggest a potential pro-fibrotic role for IL-23 in the development of chronic inflammatory autoimmune diseases characterized by intense fibrosis.
- IL-23
- autoimmunity
- inflammation
- fibrosis
1. Introduction
IL-23 is a factor involved in the development of autoimmune diseases, such as multiple sclerosis (MS), rheumatoid arthritis (RA), and systemic lupus erythematosus; it carries out its activity by stimulating and activating the pathogenic Th17 cells. Therefore, IL-23 is a potential target for modulating autoimmune responses and pathogenic Th17 cell effects. Recently developed clinical trials have shown the beneficial effects of blocking the IL-23/Th17 pathway in chronic inflammatory autoimmune diseases characterized by fibrotic damage to the organs.
2. Rheumatoid Arthritis
The role of IL-23 in RA has been extensively studied in a co-morbidity that affects approximately 15% of RA patients that is termed RA interstitial lung disease (RA-ILD) [1]. How much an excessive or altered immune response mediated by Th17 activation is involved in this pathology remains to be demonstrated. Recently, insight on the direct role of IL-23 in lung fibrosis was obtained by experimentally investigating the responsiveness of lung fibroblasts to IL-23 stimulation. The induction of CCR2 expression that regulates monocyte chemotaxis and the increased fibroblast migration suggest a direct role for IL-23 in fibrotic lung disease associated with a Th17-biased immune response [2]. In this context, a process strictly correlated with fibrotic evolution [3][4] plays a role, represented by EMT, which seems to be activated in the lung by chronic inflammatory stimuli and tissue damage. The EMT process creates an environment that facilitates fibrosis when alveolar epithelial cells are injured. In this scenario, IL-23 exerts its profibrogenic role on somatic alveolar type I (ATI) epithelial cells [2]. Primary ATI cells, after prolonged culture on rigid culture dishes, clearly show signs of a gradual transformation towards a mesenchymal phenotype characterized by the loss of epithelial proteins, such as caveolin-1, and by a reorganization of the F-actin cytoskeleton, indicating the initiation of the EMT process. IL-23 appears to be actively involved in this process because the mesenchymal transformation process is accelerated by in vitro stimulation with this cytokine, which results in the loss of the epithelial marker caveolin-1 and increased expression of mesenchymal markers, such as α-smooth muscle actin (α-SMA) and collagen I/III protein. Furthermore, IL-23 significantly promotes cell migration and regulates apoptotic resistance in IL-23-transitioning-treated ATI cells [2]. IL-23-induced EMT seems to be activated and regulated by the Target of Rapamycin (mTOR)/S6 signaling pathway, which has already been demonstrated to be the pathway involved in the pro-fibrotic activity of IL-23 [5]. The hypothesis of an involvement of IL-23 in the pathogenesis of RA-ILD exerted by promoting mTOR/S6 signaling-dependent EMT in alveolar epithelial cells was supported by transcriptional sequencing analysis of human lung fibrosis biopsy tissue [2], which detected a significant increase in IL-23 mRNA expression in RA-ILD lung sections positively correlated with transitioning ATI epithelial cell [2] (Figure 1).
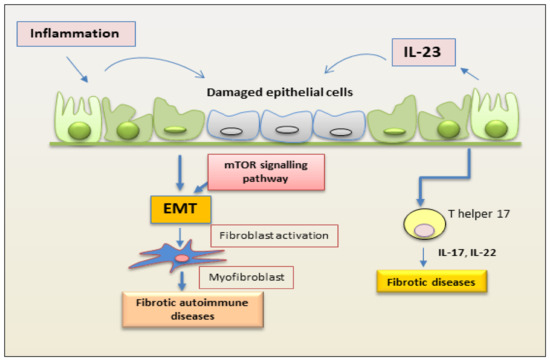
Figure 1. Schematic representation of a proposed mechanism by which IL-23 mediates EMT in epithelial cells. Inflammation promotes repetitive injury to epithelial cells that leads to the secretion of IL-23, inducing the EMT process. Injured cells start transforming into mesenchymal cells, and IL-23 amplifies the EMT process. On binding to its receptor, IL-23 activates the kinase mTOR and promotes the expression of mesenchymal markers. This process induces fibrosis in autoimmune diseases. (EMT, epithelial–mesenchymal transition; mTOR, mammalian target of rapamycin).
3. Crohn’s Disease
Intestinal fibrosis is an important complication of Crohn’s disease (CD) characterized by exaggerated proliferation of myofibroblasts and increased deposition of collagen in response to prolonged injury or chronic inflammation typical of IBD [6][7]. The mechanism underlying this hyperproliferation of myofibroblasts in CD seems to be linked, as in other pathological conditions mentioned above, to an involvement of mTOR. Experimental data report that the inhibition of mTOR determines a decreased production of IL-23, which, in turn, negatively regulates IL-22 expression and determines an improvement in the general conditions of the mouse model and a slowdown in the fibrotic evolution [7]. This inhibition of IL-23 expression is associated with elevated autophagy activity in intestinal Cx3cr1+ mononuclear phagocytes. This result paves the way to identifying a new molecular pathway that can explain the intestinal fibrotic progression in CD. The autophagy gene Atg7 knockdown determines, in fact, increased IL-23 expression and, consequently, induces the release of IL-22, triggering the fibrotic molecular events’ activation [6]. This evidence suggests, once again, a correlation between the production of IL-23 and IL-22 and identifies a new activation pathway of the fibrotic process that originates in Cx3cr1+ mononuclear phagocytes, in which the mTOR/autophagy pathway regulates the IL-23/IL-22 axis-dependent fibrosis. The synergistic action performed by IL-23 and IL-22 is elucidated and confirmed by the fact that the double inhibition of the release of both cytokines determines a decrease in all the parameters characterizing fibrosis [6] (Figure 1).
4. Autoimmune Myocarditis
Recent observations have correlated IL-23 levels with the regulation of T cells’ function in autoimmune myocarditis, thus confirming the role of IL-23 in the regulation of inflammatory processes [8]. Using mice mutated for the IL-23 gene and unable to produce a functional interleukin, IL23a−/− mice, it was demonstrated that IL-23 is necessary for the induction of cardiac inflammation in experimentally induced autoimmune myocarditis (EAM) [9]. EAM has been considered a disease characterized by the altered function of CD4+ T cells [8]. In addition, transfection experiments demonstrated that IL-23 was able to restore pathogenicity to CD4+ T cells lacking the IL-23 gene. These results support the hypothesis of a direct involvement of IL-23 in the autoimmune reactions of the heart, which could occur either thanks to the action of IL-23 on the activity of T helper cells or by inducing the secretion by the T helpers of a combination of cytokines capable of triggering autoimmune responses [10]. Both hypotheses seem to be valid and have found scientific evidence; in fact, continuous stimulation through IL-23 is necessary to determine the production of IL-17A by T helper lymphocytes. On the other hand, even a temporary stimulation by IL-23 seems to be sufficient to determine a pathogenic activation of the T helper. Confirming this, the lack of IL-23 does not compromise the establishment of an EAM condition when the T helpers have become autoreactive [9]. Based on this evidence, because high levels of IL-23 were found in patients with autoimmune myocarditis, which proceeded to stimulate T helper cells, determining an increased production and release of IL-17A, a therapy against IL-23 could be effective in blocking or delaying the fibrotic progression of the disease [9]. Moreover, elevated fibrosis and impaired heart function were detected in IL-23−/− mice but not in mice lacking the IL-12 gene at the chronic stage of the disease; this underlines the importance of IL-23-dependent T cell activation in the resolution phase of the acute stage of autoimmune myocarditis.
5. Sjögren’s Syndrome
Sjögren’s syndrome (SS) is an autoimmune disease characterized by a chronic inflammatory response that causes a morphological and functional alteration of the exocrine glands, in particular the salivary and lacrimal glands, and this seriously compromises the quality of life of these patients [3][4]. “Primary” SS is defined as a standalone entity occurring in the absence of another systemic autoimmune disease, whereas “secondary” disease is associated with the presence of other autoimmune conditions, such as RA, SLE, or systemic sclerosis (SSc). Currently, the data relating to the involvement of IL-23 in the pathogenesis of SS are still few, although very promising. Previous research revealed that IL-17/IL-23 expression was increased in mouse models of SS, highlighting that Th17 participates in lymphocytic infiltration of salivary glands and leads to lesion formation [11][12]. Another experimental study demonstrated that both protein and mRNA levels of IL-22, IL-23, and IL-17 were enhanced in the peripheral blood of patients affected by SS [13], assuming that the IL-23/IL-17/IL-22 axis could be one of the key mediators in the pathogenesis of primary SS [11][13][14][15]. Interestingly, in the absence of certain evidence of primary SS, the immunohistochemical detection of IL-17/IL-23 would classify these patients as involved in a Th17 reaction and lead to the selection of patients to be referred for subsequent periodic diagnostic screening. Based on this assumption, the use of IL-17/IL-23 immunohistochemical detection could be employed to improve the identification of SS patients with a possible diagnosis in all cases that do not fully meet the American–European criteria for pSS, in particular when the germinal center is not present at histopathological analysis and anti-SSA and anti-SSB antibodies are undetectable in the serum [16]. Recently, a role for the synergetic interaction between IL-23 and TLR was demonstrated in SS; TLR2 ligation induces the production of IL-23 and IL-17 via IL-6, STAT3, and the NF-kB pathway in primary SS. Therefore, therapeutic strategies directed against the TLR/IL-17 pathway might be valid candidates for the treatment of SS [17]. In recent years, a thriving research sector has demonstrated that SS is often accompanied by fibrotic phenomena affecting the salivary glands mediated by the activation of the EMT program, triggered by the chronic inflammation that characterizes the disease [3][4]. Therefore, based on the attribution of an important role for IL-23 in the exacerbation of the disease, there are all the premises to identify a correlation between IL-23 expression, chronic inflammation, and fibrosis in SS.
6. Systemic Sclerosis
Systemic sclerosis (SSc) is a heterogeneous chronic, autoimmune, multisystem connective tissue disorder characterized by vasculopathy, inflammation, and progressive fibrosis of the skin and internal organs [18]. Interstitial lung disease (ILD) is a major common complication, along with pulmonary arterial hypertension, which is the leading cause of morbidity and mortality in scleroderma patients [19]. The finding of pulmonary fibrosis in patients with elevated IL-23 levels had a higher frequency when compared with subjects showing normal IL-23 levels [20]. The increased release of IL-23 showed a correlation with the initial stages of the disease and with the simultaneous presence of pulmonary fibrosis; it was not associated with other clinical manifestations of SSc [20]. IL-23 has been demonstrated to be abnormally expressed in autoimmunity, including Experimental autoimmune encephalomyelitis (EAE), collagen-induced arthritis, and inflammatory bowel disease [21]. In SSc, chronic T cell activation certainly occurs, contributing to the exacerbation of tissue inflammation [18]. Recent studies imply that IL-23 may determine the differentiation of activated T cells into effector T cells in SSc [20][22]. Based on these considerations, it is possible that Th17 cells, induced by IL-23, release IL-17, which can be implicated in molecular processes leading to vascular lesions, fibrosis, and autoimmunity in patients with SSc, exploiting the binding with the IL-17 receptor expressed on fibroblasts and endothelial cells [23]. Recently, a strong correlation between the onset of the disease, the initiation of pulmonary fibrosis, and the increase of IL-23 expression has been demonstrated, suggesting that Th17, stimulated by IL-23, is involved in the onset of SSc, but not in disease progression [20]. This hypothesis has also recently found radiological confirmation because, in SSc patients, there was a statistically significant difference as regards serum concentration of IL-23 in patients with pulmonary fibrosis by chest X-ray [24].
7. Multiple Sclerosis
MS is a chronic autoimmune disorder affecting an estimated two million people worldwide. The pathological hallmarks of MS include perivascular T-cell inflammation and disseminated demyelinating lesions [25]. Experimental autoimmune encephalomyelitis (EAE) is an inflammatory autoimmune pathology that can be induced in mice and, in addition, presents many similarities with human MS [21]. EAE is a complex disease in which the interaction between several immunopathological and neuropathological events determines an approximation of the pathological characteristics of MS manifested morphologically by inflammation, demyelination, axonal loss, and gliosis [21]. The main characteristic of the EAE condition is a fibrotic scar that determines an inhibitory environment hindering the remyelination; thus, anti-fibrotic drugs may serve as novel therapeutic targets for MS. As in MS, aberrant T lymphocytes traffic against the brain and spinal cord, causing disruption of the myelin sheath integrity of the central nervous system (CNS), leading to paresthesia, paraparesis, neuritis, and ataxia [26]. Because EAE presents clinical features similar to human MS, it could be used as a model to identify the clinical efficacy of targeting the IL-23 immune pathway. Indeed, specific anti-IL-23p19 antibodies were produced to test whether blocking the functionality of IL-23 reduced the clinical symptoms of EAE and whether it could also be used in human disease [27]. The treatment of anti-IL-23p19 diminishes the serum level of IL-17 as well as the expression of IFN-γ, IP-10, IL-17, IL-6, and TNF in the CNS, thus inhibiting multiple inflammatory signaling pathways that drive CNS autoimmune inflammation. In addition, the therapeutic efficacy of the anti-IL-23p19 antibody was demonstrated to prevent disease relapse [27]. Recently, the wealth of knowledge in this field has been enriched with new discoveries that have demonstrated IL-23 as a key factor driving inflammatory processes in the CNS [28]. In particular, a transgenic mouse with astrocyte-specific expression of IL-23 developed an ataxic phenotype and cerebellar infiltrates with high amounts of B lymphocytes. In these mice, in which EAE was induced, it was demonstrated that the local IL-23 production in the CNS determines the aggravation of the disease course with severe paraparesis and an ataxic phenotype, leading to the enhancement of gliosis and neuroinflammation in the CNS [28][29]. Certainly, further studies will be necessary to identify the mechanisms that explain the key role of IL-23 in MS, but the premises are very interesting, and the preliminary results are very intriguing.
This entry is adapted from the peer-reviewed paper 10.3390/jcm12175699
References
- Laria, A.; Lurati, A.M.; Zizzo, G.; Zaccara, E.; Mazzocchi, D.; Re, K.A.; Marrazza, M.; Faggioli, P.; Mazzone, A. Interstitial Lung Disease in Rheumatoid Arthritis: A Practical Review. Front. Med. 2022, 9, 837133.
- Zhang, C.; Wang, S.; Lau, J.; Roden, A.C.; Matteson, E.L.; Sun, J.; Luo, F.; Tschumperlin, D.J.; Vassallo, R. IL-23 amplifies the epithelial-mesenchymal transition of mechanically conditioned alveolar epithelial cells in rheumatoid arthritis-associated interstitial lung disease through mTOR/S6 signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 321, L1006–L1022.
- Sisto, M.; Ribatti, D.; Lisi, S. Organ Fibrosis and Autoimmunity: The Role of Inflammation in TGFβ-Dependent EMT. Biomolecules 2021, 11, 310.
- Sisto, M.; Lisi, S. Immune and Non-Immune Inflammatory Cells Involved in Autoimmune Fibrosis: New Discoveries. J. Clin. Med. 2023, 12, 3801.
- Chen, F.; Cao, A.; Yao, S.; Evans-Marin, H.L.; Liu, H.; Wu, W.; Carlsen, E.D.; Dann, S.M.; Soong, L.; Sun, J.; et al. mTOR Mediates IL-23 Induction of Neutrophil IL-17 and IL-22 Production. J. Immunol. 2016, 196, 4390–4399.
- Mathur, R.; Alam, M.M.; Zhao, X.F.; Liao, Y.; Shen, J.; Morgan, S.; Huang, T.; Lee, H.; Lee, E.; Huang, Y.; et al. Induction of autophagy in Cx3cr1+ mononuclear cells limits IL-23/IL-22 axis-mediated intestinal fibrosis. Mucosal Immunol. 2019, 12, 612–623.
- Wang, Y.; Huang, B.; Jin, T.; Ocansey, D.K.W.; Jiang, J.; Mao, F. Intestinal Fibrosis in Inflammatory Bowel Disease and the Prospects of Mesenchymal Stem Cell Therapy. Front. Immunol. 2022, 13, 835005.
- Vdovenko, D.; Wijnen, W.J.; Zarak Crnkovic, M.; Blyszczuk, P.; Bachmann, M.; Costantino, S.; Paneni, F.; Camici, G.G.; Luescher, T.F.; Eriksson, U. IL-23 promotes T-cell mediated cardiac inflammation but protects the heart from fibrosis. Eur. Heart J. 2020, 41, ehaa946.3725.
- Wu, L.; Diny, N.L.; Ong, S.; Barin, J.G.; Hou, X.; Rose, N.R.; Talor, M.V.; Čiháková, D. Pathogenic IL-23 signaling is required to initiate GM-CSF-driven autoimmune myocarditis in mice. Eur. J. Immunol. 2016, 46, 582–592.
- Burkett, P.R.; Meyer, G.; Kuchroo, V.K. Pouring fuel on the fire: Th17 cells, the environment, and autoimmunity. J. Clin. Investig. 2015, 125, 1–9.
- Nguyen, C.Q.; Hu, M.H.; Li, Y.; Stewart, C.; Peck, A.B. Salivary gland tissue expression of interleukin-23 and interleukin-17 in sjogren’s syndrome: Findings in humans and mice. Arthritis Rheum. 2008, 58, 734–743.
- Luo, D.; Chen, Y.; Zhou, N.; Li, T.; Wang, H. Blockade of Th17 response by IL-38 in primary sjogren’s syndrome. Mol. Immunol. 2020, 127, 107–111.
- Ciccia, F.; Guggino, G.; Rizzo, A.; Ferrante, A.; Raimondo, S.; Giardina, A.; Dieli, F.; Campisi, G.; Alessandro, R.; Triolo, G. Potential involvement of IL-22 and IL-22-producing cells in the inflamed salivary glands of patients with Sjogren’s syndrome. Ann. Rheum. Dis. 2012, 71, 295–301.
- Zhan, Q.; Zhang, J.; Lin, Y.; Chen, W.; Fan, X.; Zhang, D. Pathogenesis and treatment of Sjogren’s syndrome: Review and update. Front. Immunol. 2023, 14, 1127417.
- Soypaçacı, Z.; Gümüş, Z.Z.; Çakaloğlu, F.; Özmen, M.; Solmaz, D.; Gücenmez, S.; Gercik, Ö.; Akar, S. Role of the mTOR pathway in minor salivary gland changes in Sjogren’s syndrome and systemic sclerosis. Arthritis Res. Ther. 2018, 20, 170.
- Fusconi, M.; Musy, I.; Valente, D.; Maggi, E.; Priori, R.; Pecorella, I.; Mastromanno, L.; Di Cristofano, C.; Greco, A.; Armeli, F.; et al. Immunohistochemical detection of IL-17 and IL-23 improves the identification of patients with a possible diagnosis of Sjogren’s syndrome. Pathol. Res. Pract. 2020, 216, 153137.
- Kwok, S.K.; Cho, M.L.; Her, Y.M.; Oh, H.J.; Park, M.K.; Lee, S.Y.; Woo, Y.J.; Ju, J.H.; Park, K.S.; Kim, H.Y.; et al. TLR2 ligation induces the production of IL-23/IL-17 via IL-6, STAT3 and NF-kB pathway in patients with primary Sjogren’s syndrome. Arthritis Res. Ther. 2012, 14, R64.
- Truchetet, M.E.; Brembilla, N.C.; Chizzolini, C. Current Concepts on the Pathogenesis of Systemic Sclerosis. Clin. Rev. Allergy Immunol. 2023, 64, 262–283.
- Mendoza, F.A.; Allawh, T.; Jimenez, S.A. Pharmacological treatment of systemic sclerosis-associated interstitial lung disease: An updated review and current approach to patient care. Clin. Exp. Rheumatol. 2023, 41, 1704–1712.
- Komura, K.; Fujimoto, M.; Hasegawa, M.; Ogawa, F.; Hara, T.; Muroi, E.; Takehara, K.; Sato, S. Increased serum interleukin 23 in patients with systemic sclerosis. J. Rheumatol. 2008, 35, 120–125.
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106.
- Kastelein, R.A.; Hunter, C.A.; Cua, D.J. Discovery and biology of IL-23 and IL-27: Related but functionally distinct regulators of inflammation. Annu. Rev. Immunol. 2007, 25, 221–242.
- Kurasawa, K.; Hirose, K.; Sano, H.; Endo, H.; Shinkai, H.; Nawata, Y.; Takabayashi, K.; Iwamoto, I. Increased interleukin-17 production in patients with systemic sclerosis. Arthritis Rheum 2000, 43, 2455–2463.
- Hammad, G.A.; Eltanawy, R.M.; Fawzy, R.M.; Gouda, T.M.; Eltohamy, M.A. Serum interleukin 23 and its associations with interstitial lung disease and clinical manifestations of scleroderma. Egypt. J. Bronchol. 2018, 12, 69–75.
- Khan, Z.; Gupta, G.D.; Mehan, S. Cellular and Molecular Evidence of Multiple Sclerosis Diagnosis and Treatment Challenges. J. Clin. Med. 2023, 12, 4274.
- El Behi, M.; Dubucquoi, S.; Lefranc, D.; Zéphir, H.; De Seze, J.; Vermersch, P.; Prin, L. New insights into cell responses involved in experimental autoimmune encephalomyelitis and multiple sclerosis. Immunol Lett. 2005, 96, 11–26.
- Chen, Y.; Langrish, C.L.; McKenzie, B.; Joyce-Shaikh, B.; Stumhofer, J.S.; McClanahan, T.; Blumenschein, W.; Churakovsa, T.; Low, J.; Presta, L.; et al. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J. Clin. Investig. 2006, 116, 1317–1326.
- Cua, D.J.; Sherlock, J.; Chen, Y.; Murphy, C.A.; Joyce, B.; Seymour, B.; Lucian, L.; To, W.; Kwan, S.; Churakova, T.; et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 2003, 421, 744–748.
- Ahern, P.P.; Schiering, C.; Buonocore, S.; McGeachy, M.J.; Cua, D.J.; Maloy, K.J.; Powrie, F. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity 2010, 33, 279–288.
This entry is offline, you can click here to edit this entry!