Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
Prostate cancer was the second most common cancer and ranked fifth in causing cancer-related death in men worldwide in 2020.
- prostate cancer
- DNA damage repair
1. Introduction
Similar to other cancers, epigenetic and somatic or germline genetic modifications lead to higher risk of prostate cancer and its progression [2,3,4,5]. DNA damage is implicated in carcinogenesis and may occur spontaneously or secondary to endogenous sources such as reactive oxygen species and exogenous sources including ionising radiation, chemicals or toxins, and ultraviolet (UV) radiation [6,7,8]. DNA damage response (DDR) pathways are in place to maintain genomic stability by monitoring DNA integrity and activating the DNA repair process or induce cell apoptosis if necessary [9,10,11]. Impaired DDR pathways lead to genomic instability through survival and proliferation of unrepaired cells and subsequently tumorigenesis [12]. Among germline and somatic mutations in prostate cancer, DDR defects represent 25% of them—of these, BRCA mutations are the most frequent mutation to occur [13]. BRCA1/2 genes are located at chromosome 17q21 and 13q12, respectively. They are large genes consisting of 100 and 70 kb, respectively. They have an autosomal dominant inheritance pattern with incomplete penetrance. There are multiple, major DDR pathways which are active during different phases of the cell cycle, and these include base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), homologous recombination (HR) and non-homologous end joining (NHEJ) [14]. In the case of impairment of HR, synthetic lethality induced by poly (ADP-ribose) polymerase (PARP) inhibition occurs and may target tumor tissue selectively.
2. DNA Repair Pathways
2.1. Base Excision Repair (BER)
BER is critical for repair of small base lesions that do not distort the double DNA helix caused by oxidation, methylation and deamination [15,16]. In the nucleus, it is usually prominent in the G1 phase of the cell cycle [17]. BER is initiated by one of 11 DNA glycosylases to remove the damaged base lesion and create an abasic or apurinic/apyrimidinic (AP) site [14,18]. At this site, an AP-site specific AP endonuclease (APE1) incises the DNA backbone and either of two sub-pathways occur: the missing nucleotide is inserted by DNA polymerase β (POLβ) in a process called short-patch BER (the most dominant pathway usually), or 2–13 nucleotides are replaced by a variety of proteins in the long-patch repair pathway [19]. The replaced nucleotides are then sealed by a DNA ligase [18].
Kuasne et al. found mutations in BER repair gene APEX1 have been implicated as increasing prostate cancer risk (OR = 1.68 95% CI 1.10–2.58) [20]. Furthermore, the XRCC1 gene, responsible for bringing together DNA repair proteins such as DNA ligase 3 and DNA polymerase β, has also been linked to increasing prostate cancer risk [21].
2.2. Nucleotide Excision Repair (NER)
In contrast to BER, the NER pathway repairs bulky DNA adducts and lesions that may distort the DNA helix due to UV radiation, chemotherapeutic agents such as cisplatin and environmental mutagens such as benzo[a]pyrene [14,22]. There are two sub-pathways in NER called global-genome NER (GG-NER) and transcription-coupled NER (TC-NER) [14]. The GG-NER scans the entire genome including areas that are transcriptionally inactive, whereas in the TC-NER, RNA polymerase II will stall at the lesion on the transcribed active genes [23]. In GG-NER, recognition of DNA damage is primarily through a complex of xeroderma pigmentosum, complementation group C (XPC) protein, UV excision repair protein Radiation sensitive 23B (RAD23B) protein and Centrin 2 (CETN2), which detects single-stranded DNA [24,25]. Furthermore, the ultraviolet-damaged DNA damage-binding protein (UV–DDB) complex will bind to lesions secondary to UV damage and also allow binding from downstream repair proteins such as XPC [23]. Once there is recognition of damage by the XPC-RAD23B complex, it then binds to the 10-subunit transcription initiation factor II H (TFIIH) complex to open up the DNA, track for the lesion and then incise it [22]. DNA polymerases will then fill up the repair patch and be sealed by DNA ligases [23]. In the TC-NER pathway, RNA polymerase II will identify the lesion and recruit specific proteins Cockayne syndrome WD repeat protein A (CSA) and Cockayne syndrome protein B (CSB), which subsequently recruit other core proteins [26]. The complex translocates RNA polymerase II in reverse and therefore exposes the lesion; TFIIH will bind to the site and start the chain of events to remove the lesion as described for the GG-NER pathway [27].
Mandal et al. investigated the association between NER genes XPC PAT and XPC exon 15 and prostate cancer [28]. They found a significant association between XPC PAT Ins/Ins (I/I) genotype with a 2.5-fold risk of prostate cancer (Adjusted OR- 2.55, 95% CI-1.22–5.33, p = 0.012). Similarly, the XPC exon 15 variant CC genotype showed a 2.1-fold increased risk of prostate cancer (Adjusted OR- 2.15, 95% CI-1.09–4.23, p = 0.026). The Gleason grade categorises patients with prostate cancer based on cell differentiation; the XPC PAT I/I genotype displayed a 2.8-fold increased risk of a high Gleason grade (Adjusted OR-2.88, 95% CI 1.22–6.79, p = 0.015). These results highlight how variant mutations in NER genes can increase prostate cancer risk.
2.3. Mismatch Repair
Base mismatches and insertion-deletion loops (IDLs) that occur during replication are repaired by the MMR pathway [29,30]. There are eight genes which code for the MMR pathway: hMSH2, hMSH3, hMSH5, hMSH6, hMLH1, hPMS1 (hMLH2), hMLH3, hPMS2 (hMLH4) [31,32,33]. The protein of the hMSH2 gene creates two separate heterodimers with MSH6 or MSH3; these complexes MSH2-MSH6 (MutSα) and MSH2-MSH3 (MutSβ) identify and bind to the mismatches [34]. The MSH2-MSH6 complex identifies single base mismatches and up to two nucleotide IDLs, in contrast to the MSH2-MSH3 complex which identifies up to 13 nucleotide IDLs [35]. The MutSα or MutSβ recruits the MutL complexes—the most important of which is MutLα (MLH1/PMS2 heterodimer)—which act as a mediator for other proteins to remove the mismatch; these are proliferating cell nuclear antigen (PCNA), replication factor C (RFC) and exonuclease 1 (EXO1) [14,36,37]. In particular, the EXO1 protein removes the DNA towards and past the mismatch site; once excised, MutLα suppresses EXO1 activity [36]. Finally, a DNA polymerase will synthesise DNA to replace the excision and this will be sealed by DNA ligases [38].
MMR deficiency has a prevalence of 3–12% in prostate cancer [39]. Graham et al. identified 27 men with MMR deficiency metastatic prostate cancer—the MSH2 gene was the most frequently mutated (20 men, 74%). 13 men (48%) had M1 metastatic disease on diagnosis and 19 out of 24 men (79%) who had a prostate biopsy had a Gleason score ≥ 8. They highlighted that MMR deficiency is associated with a high Gleason score and advanced metastatic disease on diagnosis, but further studies with a higher sample size are needed to investigate this further [39].
2.4. Homologous Recombination and Non-Homologous End Joining
DNA double-strand breaks (DSBs) are repaired predominantly through HR and NHEJ [40]. HR occurs in the S and G2 phases of the cell cycle as it requires a template of a sister chromatid and will repair the DNA damage error-free; compared to NHEJ which occurs throughout the cell cycle, but especially in the G1 phase and is error-prone as it ligates the ends of broken DNA without a template [4,41]. In NHEJ, both free ends of DSBs will be recognized and bound by the Ku70/80 heterodimer and this recruits the DNA-PK catalytic subunit (PKcs) to form a multi-unit complex [42]. This complex then recruits further proteins such as Artemis and PNK to repair into a normal DNA structure, polymerases to bind the broken ends, and ligases to seal both DNA strands [43,44,45,46]. The NHEJ process is error-prone without using a DNA template and can consequently drive genomic instability [4]. In contrast, HR is predominantly error-free due to identical sister chromatids being present [47]. Damaged DNA will be identified and the MRN complex (MRE11-NBS1-RAD50) will be recruited, which will activate ATM and the RAD3-related ATR kinase [48]. This will stop the cell cycle to allow DNA repair to occur and create 3′-single-stranded DNA (ssDNA) ends [47]. The exposed ssDNA will be coated by Replication Protein A (RPA), which will be replaced by Rad51 in a BRCA1/2 dependent pathway to complete the recombinase reaction [49]. The BRCA1/2 genes are key mediators in the HR pathway and any BRCA mutations would impair this HR pathway [4,50]. For example, BRCA1/2 mutations can lead to HR deficiency which would allow tumorigenesis [51]. Figure 1 provides an overview of the different DSB repair pathways.
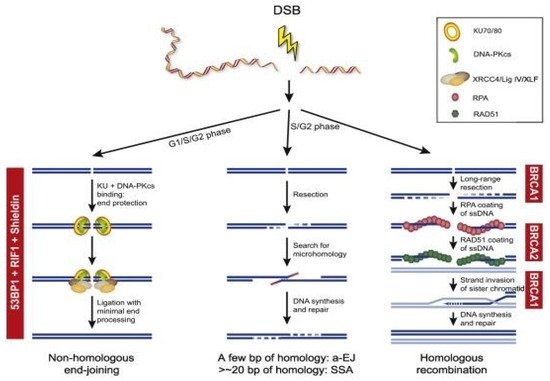
Figure 1. Overview of the different DSB repair pathways. End resection is the essential step for the choice between cNHEJ and HR, a-EJ, and SSA. The availability of a sister chromatid will direct HR. Different steps during HR are dependent on BRCA1 and BRCA2. NHEJ is dependent on the activity of 53BP1, RIF1, and the Shieldin complex (REV7, SHLD1, SHLD2, and SHLD3).
Germline BRCA mutations have been linked to reduced metastasis-free survival in localized prostate cancer and the importance of detecting these mutations will be discussed below [52].
2.5. Synthetic Lethality
Synthetic lethality is a phenomenon where inactivation of one gene allele allows cell survival compared to cell death when multiple gene alleles are disrupted [53]. Cancer cells suffer from increased oxidative stress levels and subsequent DNA damage [50,54]. DDR pathways would usually repair this DNA damage, but DDR inhibitors have been used to achieve synthetic lethality that leads to tumor cell apoptosis [50]. An example of this are the PARP inhibitors which have been used in treatment of prostate cancer [55]. PARP enzymes repair single-strand breaks and can also be activated in the HR repair (HRR) pathway for DSBs [53]. Therefore, PARP inhibitors lead to accumulation of single-strand DNA breaks along with DSBs, which would physiologically be repaired through the HRR pathway with BRCA1/2 and PARP [53]. However, in BRCA mutated prostate cancer, PARP inhibition would prevent tumor DNA repair and lead to tumor cell apoptosis [53]. Therefore, inactivation of both demonstrates synthetic lethality where tumor cells can no longer survive. Synthetic lethality could also be used in tumors which share molecular features of BRCA mutated tumors—known as “BRCAness” [56]. Therefore, mutation of genes other than BRCA in the HR pathway allows for greater therapeutic use of PARP inhibitors and there is ongoing research into this broader use of synthetic lethality targeting the HR pathway [57].
In summary, there are multiple DDR pathways which protect genome stability. Mutations can occur in these DDR genes and increase susceptibility to prostate cancer. The most common mutation is of the BRCA genes, which are a vital part of the HR pathway and can lead to more aggressive disease. Therefore, there has been a focus on BRCA-mutated prostate cancer in the research. Treatment for BRCA-mutated prostate cancer includes PARP inhibitors, which use the phenomenon of synthetic lethality to increase single-strand breaks and DSBs of tumor DNA—which cannot be repaired through a deficient HR or PARP inhibition—and therefore cancer cell death [58]. Their use has also expanded to target prostate cancer with non-BRCA mutated genes, but have similarity to the BRCA mutated genes [57].
3. Genomic Analysis of Prostate Cancer
Prostate cancer has a high inheritable component and genomic analysis allows identification of a mutation in screening high-risk individuals along with driving treatment decisions with specific therapeutic agents [59]. The Cancer Genome Atlas (TCGA) found molecular heterogeneity after analysis of 333 primary prostate cancer specimens [60]. Inactivation of DDR genes were found in 19% of localized prostate cancers with the majority in the HR pathway—these included BRCA1 (1%), BRCA2 (3%), RAD51C (3%). Furthermore, other vital kinases involved in DNA repair were inactivated: CDK12 (2%), ATM (4%), FANCD2 (7%) [60]. Other studies have compared the mutations found in primary and metastatic prostate cancer. Pritchard et al. analyzed germline mutations of 20 DNA repair genes in 692 men with metastatic castration-resistant prostate cancer (mCRPC) and no family history; 84 deleterious mutations in these DNA repair genes were identified in 82 men (11.8%) with the most frequent being BRCA2 at 5.3% [61]. In comparison, the Cancer Genome Atlas prostate cancer study found 4.6% of men with localized prostate cancer had germline mutations in these 20 DNA repair genes [16,61].
Grasso et al. also compared 50 mCRPC with 11 high-grade localized prostate cancer [62]. They found higher mutations in DNA repair factors such as BRCA2, ATM and RAD50 in mCRPC (46%) compared to localized prostate cancer (27%) [61]. Furthermore, the study by the International Stand Up to Cancer/Prostate Cancer Foundation team (SU2C-PCF) analyzed 150 metastatic prostate cancer specimens and identified 8% with germline DDR mutations and 23% with somatic DDR alterations [63]. Of the samples, BRCA2 was the most frequently mutated (13%), followed by ATM (7.3%), MSH2 (2%) and BRCA1, FANCA, MLH1, RAD51B and RAD51C (0.3% for all) [61]. Germline BRCA2 mutations were identified in 5.3%, which is higher than observed in primary prostate cancer [16,61]. PROREPAIR-B was the first prospective study to determine the prognostic impact of BRCA2 as well as BRCA1, ATM and PALB2 on cause-specific survival (CSS) in 419 mCRPC patients [64]. The study did not reach the primary end-point as there was no statistically significant difference in CCS between non-carriers and carriers of ATM, BRCA1/2 and PALB2 (33.2 months vs. 23.3 months, p = 0.264). However, germline BRCA2 mutation was identified to be a negative prognostic factor on CSS at 17.4 months compared to 33.2 months in non-carriers (p = 0.027). Therefore, BRCA2 mutation was confirmed as an independent prognostic factor for CSS (p = 0.033) [64].
These studies demonstrate both germline and somatic mutations, especially in DDR genes, are prevalent in both primary and metastatic prostate cancer. Testing for these mutations early on could identify those who may progress to advanced metastatic disease as well as specific individualized treatment options [65]. Germline mutation testing allows screening in men who are of risk of prostate cancer, while somatic mutation testing can determine treatment options [66].
The advent of PARP inhibitors in mCRPC has popularized germline testing of mutations in DNA repair genes such as BRCA1 and BRCA2 [67]. Guidelines for the above have been framed as a necessity in an era of precision oncology. The National Comprehensive Cancer Network (NCCN) Prostate Cancer Guideline Version 4.2019 recommends germline testing for BRCA1, BRCA2, PALB2, ATM, CHEK2, FANCA, RAD51D and HOXB13. Criteria include risk group, pathology and family history. All metastatic PC under the high or very high risk NCCN groups warrant testing. Low to intermediate risk groups are tested in the presence of intraductal pathology or a background of suspicious family history. This implies males in the family diagnosed with PC less than 60 years of age or those who died from it, Ashkenazi Jewish ancestry, or having at least three cancers running in the family consistent with hereditary breast ovarian cancer (HOBC) or Lynch syndrome [68]. The NCCN Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic Guideline (Version 1.2020) recommend testing specifically for high penetrance genes including BRCA1 and BRCA2. Criteria include men with: (1) metastatic prostate cancer or intraductal pathology; (2) Gleason ≥7 of Ashkenazi Jewish ancestry; (3) ≥1 first, second or third degree with breast cancer diagnosed ≤50 years of age or ovarian/pancreatic/metastatic prostate cancer/intraductal prostate cancer diagnosed at any age; (4) ≥2 first, second or third degree relative with breast cancer or prostate cancer irrespective of Gleason and age of diagnosis [69].
The Philadelphia Prostate Cancer Consensus Conference 2019 was an international effort to gain a multi-disciplinary consensus among oncology, urology, cancer genetics, epidemiology, patient advocates, and NCCN leaders to arrive at uniform guidelines for germline testing. It was recommended for men with mCRPC and castration-sensitive prostate cancer and for men with strong family history of two or more male relatives diagnosed with prostate cancer at <60 years of age or died from the same. Additional considerations for testing included pathologic criteria (intraductal pathology; advanced disease of T3a or higher; or International Society of Urological Pathology [ISUP] Grade Group 4 or above) and family history criteria (two or more cancers in the HBOC or Lynch syndrome spectrum diagnosed at <50 years of age or having Ashkenazi Jewish ancestry) [70]. The NCCN Prostate Cancer Early Detection Guideline Version 2.2019 recommends considering BRCA1/2 status for prostate cancer screening starting at 40 years of age or to consider annual screening vs every two-year screening among BRCA1/2 mutation carriers [68]. The NCCN Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic Guideline Version 1.2020 recommends Prostate Specific Antigen (PSA) screening start at 40 years of age for BRCA1 and BRCA2 carriers [69]. The 2019 Philadelphia Prostate Cancer Consensus Conference recommended that BRCA2 mutation status be a part of prostate cancer screening discussions and to start prostate cancer screening at age 40 or 10 years prior to the youngest prostate cancer diagnosed in a first-, second- or third-degree male relative [70].
The European Society for Medical Oncology (ESMO) Clinical Practice Guidelines for prostate cancer recommend tumor testing for homologous recombination genes and mismatch repair defects (or microsatellite instability) in all patients with mCRPC. Patients with pathogenic mutations in cancer-risk genes identified through tumor testing should be referred for germline testing and genetic counselling. Germline testing for BRCA2 and other DDR genes associated with cancer predisposition syndromes is warranted in patients with a family history of cancer [71]. The European Association of Urology-European Association of Nuclear Medicine-European Society for Radiotherapy and Oncology-European Association of Urology Section of Urological Research-International Society of Geriatric Oncology (EAU-EANM-ESTRO-ESUR-ISUP-SIOG) guidelines also recommends germline testing in metastatic prostate cancer. Their criteria include—men with (1) high-risk prostate cancer with a relative diagnosed with prostate cancer at age < 60 years; (2) multiple relatives diagnosed with prostate cancer at age < 60 years or died from it; (3) family history of high-risk germline mutations or multiple cancers on same side of family. However, the strength of these recommendations is “weak” [72].
Contrary to the above advocacy of germline testing all around the globe, the United Kingdom’s National Institute for Health and Care Excellence (NICE) does not recommend the PARP inhibitor olaparib for the treatment of patients with hormone-relapsed, metastatic prostate cancer that harbour BRCA1 or BRCA2 mutations that have progressed on abiraterone or enzalutamide. According to them, treatment options for the above cohort include docetaxel, cabazitaxel or radium-223. Although clinical trial data have demonstrated that patients who received olaparib have extended progression-free survival (PFS) and overall survival (OS) over those who received re-treatment with either abiraterone or enzalutamide, evidence is unclear because re-treatment with those agents is not considered to be effective and is not standard of care in the National Health Service (NHS). It is also uncertain how effective olaparib is compared with cabazitaxel, radium-223 or docetaxel because there is no evidence directly comparing them. Hence, NICE is one of the very few which have no guidance on germline testing of prostate cancer [73].
This entry is adapted from the peer-reviewed paper 10.3390/ijms222312628
This entry is offline, you can click here to edit this entry!