1. Introduction
In 1886, the German chemist Ernst Schulze and his assistant Ernst Steiger extracted arginine from yellow lupine germ buds for the first time. In 1899, Schulze and Winterstein synthesized arginine (L-Arg) from L-ornithine and cyanamide, and, in 1910, the L-Arg structure was confirmed by Sorensen [
1]. In 1987, nearly 100 years later, arginine-derived nitric oxide (NO) was shown to be a factor regulating vascular tone. NO was classified as a physiologically active intermediate product of L-Arg to nitrite/nitrate conversion in macrophages and endothelial cells. The discovery of the fundamental role of NO-related compounds in the physiology of the human cardiovascular system resulted in the 1998 Nobel Prize being awarded to Robert F. Furchgott, Luis J. Ignarro and Ferid Murad [
2].
L-Arg is a dibasic cationic amino acid involved in various metabolic pathways [
3,
4]. L-Arg is mainly derived from the three sources in the in vivo–dietary intake, endogenous de novo production from L-citrulline, or protein catabolism. Endogenous L-Arg is largely synthesized in the renal proximal tubules from L-citrulline generated from dietary L-Arg, in intestinal epithelium [
5]. Plasma L-Arg concentrations range from 50 to 250 µM [
4,
6,
7,
8,
9], which is much lower than in subcellular compartments, where its concentration level reaches 1 mM [
10].
Evidence suggests L-Arg is an mTOR pathway-signaling molecule [
11]. There are at least five arginine sensors (GPRC6A, SLC38A9, CASTOR1, CASTOR2, TM4SF5) reported in the literature [
12,
13,
14,
15]. mTOR is considered the key regulator of important cellular processes, including protein synthesis, proliferation, autophagy, lysosomal function, metabolism and inflammation. Research into the influence of L-Arg on mTOR regulation opens a new intriguing chapter in L-Arg investigation history [
16].
In mammalian cells, at least eight transporters are involved in L-Arg trafficking across the plasma membrane [
17]. L-Arg is metabolized by those four groups of enzymes: arginases, nitric oxide synthases (NOS), arginine decarboxylase (ADC) and arginine:glycinamidinotransferase (AGAT). Among them, arginases and NOS exist in several different isoforms.
Arginases are manganese-containing enzymes hydrolyzing L-Arg into L-ornithine and urea in the liver-urea cycle important for ammonia detoxification. L-ornithine is a substrate for ornithine decarboxylase (ODC) that initiates the synthesis of polyamines. Alternatively, ornithine aminotransferase (OAT) can metabolize L-ornithine into proline. These metabolites are involved in regeneration processes. Polyamines (putrescine, spermine and spermidine) are necessary for cell proliferation, whereas proline enriches collagen. It was established that arginase is expressed in many cell types, and its two isoforms catalyze the same biochemical reaction. Human arginase 1 (ARG1) is a cytosolic protein expressed mainly in hepatocytes, as well as in myeloid lineage cells. Human arginase 2 (ARG2), localized in mitochondria, widely expresses in extrahepatic tissues. Both enzymes share as low as 58% amino acid sequence homology, bearing a nearly identical structure within the catalytic site [
18,
19].
NOS metabolizes L-Arg to produce L-citrulline and NO. NOS isoforms differ in structure and mechanisms of activity regulation. Neuronal (nNOS) and endothelial NOS (eNOS) are constitutively expressed in non-immune cells: neurons, muscle, and endothelial cells. Their activity is regulated by Ca-dependent calmodulin binding as well as protein phosphorylation/dephosphorylation at the serine residue [
20,
21]. The enzymes usually produce NO in low concentration, which acts as an intracelullar signaling molecule [
22] and regulates vascular homeostasis [
23]. Inducible NOS (iNOS) expression has long been considered specific to immune cells, but recent studies show that the spectrum of the cells expressing iNOS is much broader [
23] than earlier thought. Immune cells produce NO along with reactive oxygen species (ROS) to eliminate pathogens and tumor cells. NO and its derivatives (Reactive Nitrogen Species—RNS) act at micromolar concentrations nonspecifically on various targets, which may result in normal cell damage. Therefore, iNOS activity is highly regulated. Expression of iNOS induces in response to pro-inflammatory cues: bacterial toxins, as well as cytokines interleukin (IL)-1β, interferon γ (IFNγ), and tumor necrosis factor α (TNFα). On the contrary, IL-4 and IL-10, transforming growth factor β (TGFβ), downmodulate iNOS gene expression. Mechanisms regulating the bioavailability of intracellular L-Arg play an important role in regulating NOS-mediated NO synthesis [
20,
21].
ADC and AGAT are not involved in regulating immune cell functions. In mammals, ADC is highly expressed in brain tissues [
24], whereas AGAT is found in the brain and heart [
25,
26,
27]. ADC metabolizes L-Arg into agmatine, which is, in turn, converted by agmatinase into putrescine and urea [
28]. Except for agmatine inhibitory effects on macrophage iNOS, little is known about the role of these enzymes in immune system [
29].
L-Arg plays a crucial role in detoxification of ammonia—a protein breakdown product acts as a secretagogue and serves as a substrate for the synthesis of NO, an important signaling molecule that regulates vascular tone and cytotoxic functions of macrophages. L-Arg is also a precursor in the synthesis of L-ornithine and agmatine, creatine and polyamines. Metabolism of L-Arg is involved in immune cell regulation [
30]. It is now clear that L-Arg metabolism is engaged in the pathogenesis of tumor growth, inflammation, infectious diseases, and fibrotic processes [
31,
32,
33,
34,
35,
36,
37], as well as physiological immunodeficiencies in newborns and pregnant women [
38].
2. L-Arginine Metabolism in Cancer
Being a proteinogenic amino acid, L-Arg is crucial for protein synthesis in actively proliferating cells, such as tumor cells [
59,
60,
61] (
Figure 1). L-Arg is also involved in other metabolic processes related to tumor growth, including production of NO, polyamines, nucleotides, proline, and glutamate. Moreover, it was shown that several enzymes of L-Arg metabolism (ODC) and amino acid transporters (CAT and SLC6A14) are actively involved in developing tumors [
62]. In contrast, it is known that immune cell activity also dramatically relies on L-Arg bioavailability [
39,
63]. Therefore, L-Arg metabolism has an ambiguous role in oncology.
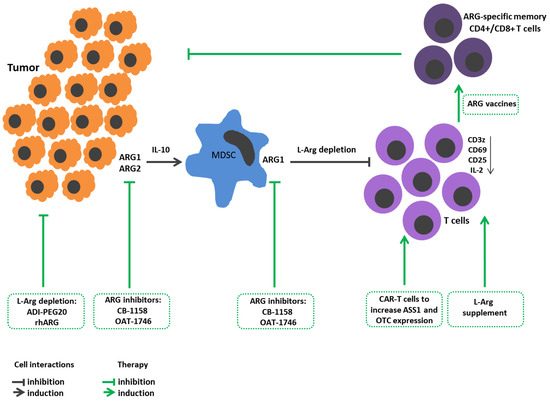
Figure 1. L-Arginine metabolism in tumor growth immunopathogenesis and arginine-dependent processes as a target of therapeutic approaches. Expression of arginases by tumor cells promotes their proliferation and metastasis. Arginase-mediated depletion of L-Arg in the tumor microenvironment contributes to the development of immunosuppression. Tumors also induce arginase in the microenvironment. Tumor-infiltrating arginase-expressing MDSC metabolites stimulate tumor mutagenesis, reinforce immunosuppression, decrease CD3z, CD25, CD69 expressions in T cells and IL-2 production. Therapeutic strategies: Arginine-hydrolyzing enzymes result in L-Arg exhaustion in tumor microenviroment and inhibit tumor cell proliferation. Administration of ARG inhibitors decreases tumor growth/metastasis. L-Arg supplement restores T cell functions. Reengineering CAR-T cells increases ASS1 and OTC expression to improve T cell L-arg bioavailability. Elaboration ARG1-specific CD4+ and CD8+ T cell promotes arginase-positive cell elimination. ADI-PEG20, pegylated arginine deiminase; ARG1, arginase 1; ARG2, arginase 2; ASS1, argininosuccinate synthase; CAR-T, chimeric antigen receptor T cells; L-arg, L-arginine; MDSCs, myeloid derived suppressor cells; OTC, ornithine transcarbamylase; rhARG, recombinant human arginase.
Tumor cells require arginases for better proliferation and metastasis. For instance, ARG1 in neuroblastoma cells triggers expression of AKT and ERK signaling cascades leading to cell proliferation [
64]. ARG2 in thyroid tumors elevates expression of proliferative markers, such as Ki-67, PCNA [
65], whereas, in melanoma, it contributes to a promigratory cell phenotype with a high level of adhesion molecule ICAM-1 [
66]. Patients with higher ARG1 expression showed less aggressiveness, invasiveness, metastasis, and higher differentiation of hepatocellular cancer cells [
67]. Some studies report that high ARG2 expression in squamous cell carcinoma of the head and neck is associated with a poorer outcome [
68].
At the same time, L-Arg deprivation during tumor growth becomes one of the mechanisms for reprogramming immune responses with induced immunosuppression. Therefore, arginase-expressing tumor cells can simultaneously solve two issues, on the one hand, generating metabolites necessary for growth, and, on the other hand, reprogramming the tumor microenvironment to suppress the immune reactions.
Some tumors implement these strategies by inducing arginase in their microenvironment [
69,
70]. That was supported in the study by Lian et al. showing that colorectal cancer cells co-cultured with monocytic myeloid leukemia cells (THP-1) with upregulated ARG1 expression in the latter, and that colorectal cancer cells contributed to monocyte differentiation towards the anti-inflammatory M2 cells secreting IL-10 [
71]. Tumor-infiltrating arginase-expressing monocyte lineage cells can induce tumor immunoresistance [
70]. A high expression of ARG1 is found in peripheral blood myelocytes of patients with breast cancer [
72].
In the late 1990s, myeloid lineage cells in the tumor microenvironment were described. They differed from macrophages, had immature neutrophil and monocyte morphology, and exhibited a strong suppressive activity against T cells. These cells were dubbed myeloid-derived suppressor cells (MDSCs). It is now recognized that MDSCs comprise two main well-defined subsets: mononuclear (M-MDSC) and polymorphonuclear (PMN-MDSC) MDSCs. Paulo C. Rodriguez et al. characterized such cells as mature myeloid cells with high arginase activity [
73]. Murine studies demonstrated that inhibition of arginase with Nor-NOHA exerted antitumor effects in vivo. MDSCs, along with arginase, possess multiple mechanisms to regulate immune cell function, including production of NO, peroxynitrite, superoxide and hydroxyl peroxide, prostaglandin E2, TGFβ, and adenosine [
74]. The accumulation of MDSCs is associated with a negative clinical outcome in cancer patients, as well as a poor response to various immunotherapeutic strategies [
74]. Low plasma L-Arg levels were linked to increased surface expression of the inhibitory costimulatory molecule PD-L1 on myeloid lineage leukocytes and worse cancer survival [
75]. It is suggested that MDSCs migrating to the lymph nodes may restrain T cell activation and clonal expansion and thereby terminate immune response during infection [
76]. MDSCs are also considered an essential player in regulating immune response in chronic inflammation, trauma and autoimmune diseases [
74].
L-Arg is necessary for lymphocyte activation and proliferation, as well as execution of related effector functions during immune response. Availability of L-Arg ex vivo within a physiological range modulates CD3z expression levels [
39,
77,
78], using a mechanism not yet fully elucidated [
4]. The regulation of CD3z expression is a crucial mechanism for modulating T cell activation. L-Arg deprivation affects TCR signaling by reducing F-actin level and disrupting the immune synapse structure in activated T cells [
79]. L-Arg depletion also prevents nuclear translocation of NF-κB (nuclear factor κB) p65 [
80]. L-Arg deficiency lowers IL-2 production and the expression of early activation markers CD25 and CD69 in cultured human T cells [
81]. A sufficient amount of L-Arg is a necessary condition for T lymphocyte entry into the cell cycle [
82]. L-Arg demand markedly elevates upon T cell activation, and the amino acid supplement promotes CD4+ and CD8+ T cell survival and antitumor activity [
83]. It was shown that, in the absence of L-Arg, NK cell granule exocytosis and cytotoxicity are completely abrogated, and cytokine secretion and proliferation are profoundly suppressed [
84]. A high expression of arginases in a tumor microenvironment suppresses T cell response. A tumor microenvironment deficient in L-Arg contributes to developing cytopenia because this amino acid is required for proliferation of myeloid and CD34+ hematopoietic cells [
70].
Two opposing strategies are used in tumor therapy targeting L-Arg metabolism. One strategy is to increase L-Arg bioavailability to immune cells for stimulating an antitumor immune response [
85]. L-Arg added at supraphysiological concentration stimulates antitumor activity of CD8+ T cells not only in a cell culture, but also in vivo upon reinfusion into tumor-bearing mice [
83]. Remarkably, T cells encountering L-Arg deficiency deploy mechanisms to maintain the intracellular amino acid reserves [
86] by upregulating ASS1 for resynthesis of L-Arg from L-citrulline [
87]. Therefore, reengineering chimeric antigen receptor T cells (CAR-T) to increase ASS1 and ornithine transcarbamylase (OTC) expression promotes persistence of CAR-T cells in vivo and their activity against solid and hematological tumors [
88]. Hence, CAR-T therapy combined with enhanced L-Arg bioavailability can increase antitumor therapeutic efficacy. Introducing L-Arg supplementation may also potentiate M1 macrophage antitumor activity, because L-Arg is the sole substrate in the iNOS-mediated NO generation [
89].
Recently, new drugs—arginase inhibitors—have been tested. Arginase inhibitor-substance CB-1158 has performed well in experiments on mice and cell cultures. The mechanism of the drug action is aimed at blocking ARG1 in MDSCs, resulting in T lymphocyte active proliferation and increased CD8+ T cell tumor infiltration [
90]. Another drug, OAT-1746, a selective inhibitor of ARG1/ARG2, had no effect on viability but suppressed the growth of tumor cells, abrogated tumor metastasis, and enhanced the anti-PD1 antibody-related antitumor effect in mouse models and in vitro [
91].
ARG1-specific IFNγ-producing Th1 CD4+ T cells were found in the mononuclear fraction of peripheral blood in patients with melanoma [
92]. CD8+ T cells from melanoma patients also showed an immune response against ARG1. These ARG1-specific CD4+ and CD8+ T cells predominantly displayed the memory cell phenotype (CD45R0+) [
93]. Based on these data, the generation of so-called “arginase vaccines” inducing arginase-specific T cell clones is considered among the promising strategies in antitumor therapy. When ARG1 peptide vaccine was tested in mice, the data indicated enhanced immune response against tumor cells. It was also shown that, combined with PD-1 inhibitors, such vaccination induces a more potent antitumor response [
94]. In 2022, a human trial with ARG1 peptide vaccine demonstrated its safety. At the same time, nine (90%) out of ten patients had measurable peptide-specific reactions in the peripheral blood, and in two (20%) out of ten patients, the disease was stabilized during treatment [
95].
Another therapeutic approach for oncological diseases is aimed at suppressing tumor cell proliferation, metastasis, and invasion and, paradoxically, proposing L-Arg deprivation. The most common current method of amino acid deprivation is supposed to use arginine-hydrolyzing enzymes. The best studied drugs are presented by pegylated arginine deiminase (ADI-PEG20) and recombinant human arginase (rhArg).
ADI-PEG20, via L-Arg depletion, was shown to induce autophagy and subsequent cancer cell death [
96]. ADI-PEG20 worked well in stage II clinical trials in patients with hepatocellular carcinoma [
97]. Also, ADI-PEG20 was highly effective in ASS1 negative melanoma patients [
98]. The best effect was observed while using ADI-PEG20 with chloroquine [
99] or cytostatics, as they inhibit autophagy, and more tumor cells undergo apoptosis. Thus, when using ADI-PEG20 with cisplatin, a more intense apoptosis was observed in melanoma cells [
100]. The use of ADI-PEG20 has its drawbacks and limitations. Since L-citrulline is one of the metabolites of ADI, ASS1/ASL-positive tumors are resistant to therapy with this drug [
101,
102], while some ASS1-negative tumors acquire the ability to express ASS1 after ADI-PEG20 treatment [
99,
103].
Arginases, unlike ADIs, metabolize L-Arg into ornithine and uric acid, rhArg, which affects ASS1-positive tumors [
101]. A pegylated counterpart (Peg-rhARG1) showed positive results in a phase I clinical trial in patients with hepatocellular carcinoma [
104].Furthermore, rhArg coupled to an IgG1 Fc fragment (rhArg-Fc) is currently being assessed in vitro [
105]. Autophagy inhibitors, including chloroquine and bafilomycin A1, potentiate rhArg- and ADI-PEG20-induced cytotoxicity [
105].
This entry is adapted from the peer-reviewed paper 10.3390/cimb45040231