Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
The field of venous thromboembolism has undergone numerous innovations, starting from the recent discoveries on the role of biomarkers, passing through the role of metabolomics in expanding the knowledge on pathogenic mechanisms, which have opened up new therapeutic targets. A variety of studies have contributed to characterizing the metabolic phenotype that occurs in venous thromboembolism, identifying numerous pathways that are altered in this setting.
- venous thromboembolism
- deep vein thrombosis
- DVT
- D-Dimer
1. Introduction
The last few years have seen the maturation of numerous discoveries in the field of venous thromboembolism. In particular, more and more evidence has been reported regarding biomarkers, the role of metabolomics, and that of COVID-19, not to mention the advent of direct oral anticoagulants, which have rapidly established themselves among the treatment options. Deep vein thrombosis (DVT) and pulmonary embolism (PE) represent the major diseases of venous thromboembolism (VTE) [1]. DVT usually involves the deep veins of the lower or upper limbs but can occur in other sites. Occlusion of the deep veins in a limb by a thrombus damages drainage of blood, thereby leading to pain and swelling distal to the obstruction [1]. Pulmonary embolism refers to a block of a pulmonary artery by a thrombus that has traveled from elsewhere in the body, through the bloodstream, and to the lungs. DVT in the legs—or less commonly, the arms—is by far the leading source of pulmonary embolism [1]. The incidence of thromboembolism increases with increasing age. Women are usually affected at a younger age [2]. Approximately two-thirds of VTE cases are associated with deep vein thrombosis and 80% are proximal [2]. Distal (below the knee) DVTs are more transient episodes, whereas proximal DVTs are related to chronic conditions [2][3]. Deep venous thrombosis is frequently secondary to heritable and acquired risk factors [4]. Heritable risk factors include abnormalities associated with hypercoagulability of the blood, the most common of which are factor V Leiden and the prothrombin G20210A gene mutations [4]. Acquired risk factors include advanced age, history of previous VTE, obesity, and active cancer, all of which limit mobility and may be associated with hypercoagulability [5][6]. Superimposed on this background risk, VTE often occurs in the presence of triggering factors, which increase the risk above the critical threshold. In 25–50% of first episodes of DVT, no trigger is identified [7][8].
Triggering factors such as surgery, trauma, and pregnancy or estrogen therapy lead to endothelial cell activation, stasis, and hypercoagulability, which are the components of the Virchow triad [9][10]. In 25–50% of first episodes of DVT, no predisposing factor is identified [11]. The main complications involve the extension of thrombosis and the recurrence of PE and DVT [11]. Long-term complications include post-thrombotic syndrome (PTS), which is characterized by chronic venous symptoms and/or signs secondary to DVT [11]. This is the most frequent chronic complication of DVT and occurs in 30–50% of patients within 2 years of proximal DVT [11]. A previous ipsilateral DVT, proximal location (ileo-femoral > popliteal), and stenosis of residual veins are the most significant risk factors for PTS [12].
2. Biomarkers in Venous Thromboembolism
Considering that VTE can often present with few symptoms, it would be useful to know biomarkers that enable early identification of patients at high or low risk of primary and recurrent VTE. Various established and novel biomarkers associated with VTE have been investigated with regard to their potential for predicting primary or recurrent VTE, for facilitating the diagnosis of VTE, and for optimizing the clinical management of VTE. Actually, these biomarkers can be divided into two categories from the pathobiology of DVT or thrombotic disease. One is coagulation markers, such as D-dimer, Thrombin, etc. while the other is inflammatory markers, including P-selectin, inflammatory cytokines, and microparticles (Figure 1).
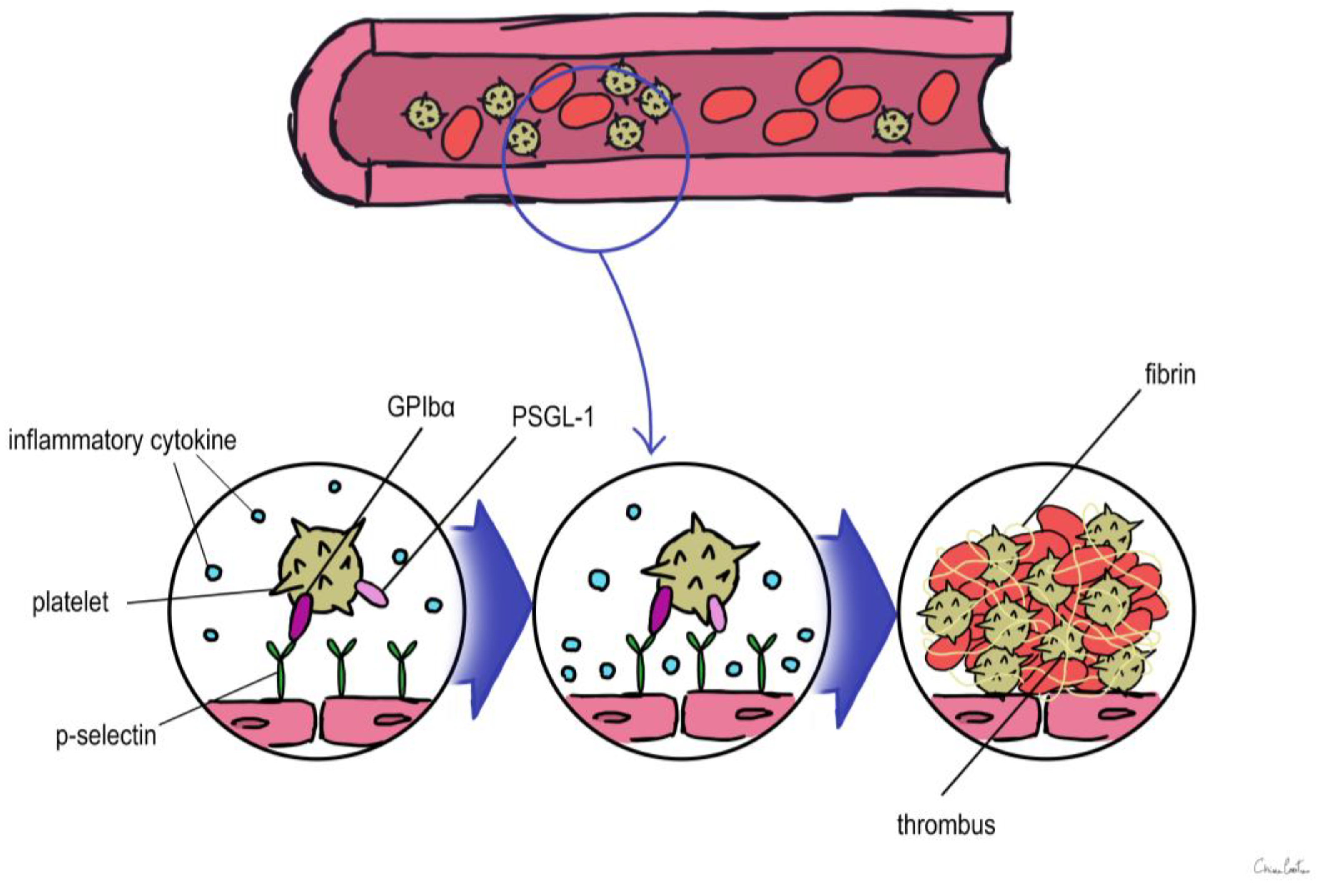
Figure 1. Overview of the role of biomarkers during thrombus formation. PSGL-1: P-selectin glycoprotein ligand 1.
2.1. D-Dimer
D-Dimer is a cross-linked fibrin degradation product that forms right after throm-bin-generated fibrin clots are broken down by plasmin and indicates a general stimulation of blood coagulation and fibrinolysis [13]. Testing for D-Dimer was investigated as a tool for the diagnosis of VTE and has been incorporated into diagnostic algorithms in the management of patients with suspected VTE since D-Dimer levels rise during a critical incident of VTE [13][14][15]. D-Dimer is the best-recognized biomarker for the first assessment of suspected VTE; a negative result of D-Dimer may confidently rule out both DVT and PE with a high sensitivity of up to 95% and a negative predictive value of almost 100% [16]. D-Dimer testing must be incorporated into thorough sequential diagnostic methodologies that involve clinical probability assessment and imaging tools because of its poor specificity for proving VTE [16]. D-Dimer was examined as a risk factor for the occurrence of a future first incident of VTE and was related with a three-fold higher risk in a population-based cohort analysis [16]. Additionally, in prospective cohort studies, D-Dimer levels are a well-researched biomarker for the estimation of the risk of VTE recurrence following the cessation of oral anticoagulant therapy [16]. In subjects with prior unprovoked VTE, Palareti et al. measured D-Dimer levels 1 month after discontinuing oral anticoagulation and found that normal levels (500 ng/mL) had a high negative predictive value for VTE recurrence [17]. In a different investigation, Eichinger et al. demonstrated that elevated D-Dimer levels were linked to an even greater risk of recurrent VTE, particularly in individuals with congenital thrombophilia, such as a factor V Leiden or prothrombin variant [18]. On the basis of the above evidence, the authors Eichinger et al. concluded that the de-termination of the duration of oral anticoagulation for secondary VTE prevention may be influenced by the measurement of D-Dimer, which has become a pillar in the diagnostic work-up of patients with suspected VTE and is essential in the identification of hyper-coagulable conditions [18].
2.2. Thrombin
Thrombin is crucial for the acceleration of the coagulation cascade because it activates platelets, Factor V, and FVIII and because it is an essential part of a positive feedback loop that causes the production of a significant amount of additional thrombin, the conversion of fibrinogen to fibrin, and ultimately the formation of clots. Some studies in the past have demonstrated that TG is one of the risk factors for VTE and can be used as a predictive marker to assess thrombosis [19][20]. Many authors across the years, such as Lutsey et al. [21] and Vilieg et al., have tried to show the lack of an association between increased TG level and the recurrence of VTE; however, some investigators thought TG parameters alone were inappropriate for the exclusion of DVT or to predict the risk of recurrence of VTE [22].
2.3. P-Selectin
P-selectin, which is stored in the granule membrane of resting platelets (a-granules) and endothelial cells (Weibel–Palade bodies) [23], is a member of the selectin family of cell adhesion molecules together with E-selectin and L-selectin [24]. The primary ligand for P-selectin in vivo is P-selectin glycoprotein ligand 1 (PSGL-1), which is expressed in the majority of leukocytes and is also present in trace levels on platelets [24]. Transmembrane P-selectin is redistributed onto the cell surface after cell activation and partially discharged into the bloodstream in its soluble form (sP-selectin) [24]. It facilitates the interaction of leukocytes that express PSGL-1 with activated platelets and endothelial cells [25][26][27]. In humans, elevated levels of soluble P-sel (sP-sel) are typical in DVT and VTE [27]. The interaction between P-selectin and PSGL-1 is crucial for thrombus development [28][29][30]. P-selectin was shown to have an impact on fibrin deposition in the thrombus by Palabrica et al. [29]. They discovered that inhibiting P-selectin interactions selectively prevented fibrin from being deposited on a thrombogenic graft in a baby as well as leukocyte adherence to platelets [29].
Korne l Miszti-Blasius et al. [30] discovered that PSGL-1-null mice had milder thrombocytopenia, less fibrin deposition, and a smaller number of thrombosed blood vessels after giving collagen with epinephrine to wild-type and PSGL-1 knockout mice. As a result, it is conceivable that a lack of PSGL-1 might prevent leukocyte–platelet interactions and lessen the likelihood of thrombus development [30]. Using sP-sel as a biomarker may improve the positive predictive value (as determined by a positive duplex ultrasound). According to a study evaluating the use of sP-sel in combination with a Wells risk prediction score for diagnosing VTE, this combination may be able to rule in the diagnosis of DVT with a sensitivity of 91% (low sP-sel and low Wells score to rule out the diagnosis) and a specificity of 98% (high sP-sel and high Wells score to rule in the diagnosis) [31].
In conclusion, a significant antithrombotic impact was seen when PSGL-1 and P-selectin interacted [31]. As a result, focusing on P-selectin or its ligand PSGL-1 may offer a viable treatment strategy for clinical circumstances. Numerous studies have revealed that the P-selectin–PSGL-1 interaction induces a procoagulant state by causing the formation of leukocyte-derived microparticles [32] and mediating the transfer of tissue factor (TF) to platelets [33]. This is in addition to its roles in mediating the binding of platelets and endothelial cells with leukocytes and enhancing fibrin deposition [33].
2.4. Inflammatory Cytokines
An increasing number of studies point to a role for inflammatory markers in VTE, including CRP and interleukin (IL)-1b, 6, 8, and 10. An initiator of the extrinsic route of coagulation, TF, may be affected by inflammatory cytokines, potentially setting off thrombotic illness [34]. Recent laboratory investigations have shown that elevated CRP levels significantly impact the development of VTE [35][36].
It is likely that mutations in genes encoding for proteins involved in inflammation may affect susceptibility to VTE based on the link between inflammation and coagulation [37]. Inflammation-related gene polymorphisms were looked at by Beckers et al. in both the VTE patient and control groups. It was discovered that IL-1A, IL-4, IL-6, and IL-13 Polymorphisms were linked to the development of VTE [38].
Several authors [39][40] have researched an association between proinflammatory cytokines and VTE. Reitsma and Rosendaal’s [39][40] case-control study not only showed an association with cytokines such as IL-1 beta, IL-6, IL-10, and TNF alpha but also revealed a deterioration of endothelial function in patients with VTE [39][40].
According to research by Downing et al. [41], exogenous IL-10 supplementation reduced inflammation and thrombus development whereas IL-10 neutralization enhanced thrombosis and inflammation. They suggested that IL-10 may be employed therapeutically to treat VTE [41].
2.5. MPs (Microparticles)
MPs, which are by definition between 0.1 and 1.0 lm in size, are small membranous vesicles released from the plasma membranes of platelets, leukocytes, red cells, and endothelial cells in response to apoptosis or cellular activation [42][43]. In the past, MPs were considered cellular debris without a biological function. In recent years, several studies have highlighted its role in inflammation and vascular function [44].
In the context of hypercoagulable conditions, elevated MP values have been found, and above all they have been highlighted in patients with VTE [45]. Moreover, MPs are the primary carriers of circulating TF, the principal initiator of intravascular thrombosis. However, at the moment, there are not enough studies (only animal studies) that clarify their role.
A new horizon in the management of VTE may be the application of metabolomics profiling in the area of vascular diseases, which can become a game changer in early diagnosis and patient management.
This entry is adapted from the peer-reviewed paper 10.3390/ijms241713411
References
- Jiang, X.; Zeleznik, O.A.; Lindström, S.; Lasky-Su, J.; Hagan, K.; Clish, C.B.; Eliassen, A.H.; Kraft, P.; Kabrhel, C. Metabolites Associated with the Risk of Incident Venous Thromboembolism: A Metabolomic Analysis. J. Am. Hear. Assoc. 2018, 7, e010317.
- Spencer, F.A.; Emery, C.; Joffe, S.W.; Pacifico, L.; Lessard, D.; Reed, G.; Gore, J.M.; Goldberg, R.J. Incidence rates, clinical profile, and outcomes of patients with venous thromboembolism. The Worcester VTE study. J. Thromb. Thrombolysis 2009, 28, 401–409.
- Wendelboe, A.M.; Raskob, G.E. Global Burden of Thrombosis. Circ. Res. 2016, 118, 1340–1347.
- Khan, F.; Rahman, A.; Carrier, M.; Kearon, C.; Weitz, J.I.; Schulman, S.; Couturaud, F.; Eichinger, S.; Kyrle, P.A.; Becattini, C.; et al. Long term risk of symptomatic recurrent venous thromboembolism after discontinuation of anticoagulant treatment for first unprovoked venous thromboembolism event: Systematic review and meta-analysis. BMJ 2019, 366, l4363.
- Braithwaite, I.; Healy, B.; Cameron, L.; Weatherall, M.; Beasley, R. Venous thromboembolism risk associated with protracted work- and computer-related seated immobility: A case-control study. JRSM Open 2016, 7, 2054270416632670.
- Baglin, T. Inherited and Acquired Risk Factors for Venous Thromboembolism. Semin. Respir. Crit. Care Med. 2012, 33, 127–137.
- Agnelli, G.; Prandoni, P.; Becattini, C.; Silingardi, M.; Taliani, M.R.; Miccio, M.; Imberti, D.; Poggio, R.; Ageno, W.; Pogliani, E.; et al. Extended Oral Anticoagulant Therapy after a First Episode of Pulmonary Embolism. Ann. Intern. Med. 2003, 139, 19–25.
- Murin, S.; Romano, P.S.; White, R.H. Comparison of outcomes after hospitalization for deep venous thrombosis or pulmo-nary embolism. Thromb. Haemost. 2002, 88, 407–414.
- Kushner, A.; West, W.P.; Pillarisetty, L.S. Virchow Triad. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: http://www.ncbi.nlm.nih.gov/books/NBK539697/ (accessed on 30 November 2022).
- Lurie, J.M.; Png, C.M.; Subramaniam, S.; Chen, S.; Chapman, E.; Aboubakr, A.; Marin, M.; Faries, P.; Ting, W. Virchow’s triad in “silent” deep vein thrombosis. J. Vasc. Surg. Venous Lymphat. Disord. 2019, 7, 640–645.
- Mazzolai, L.; Aboyans, V.; Ageno, W.; Agnelli, G.; Alatri, A.; Bauersachs, R.; A Brekelmans, M.P.; Büller, H.R.; Elias, A.; Farge, D.; et al. Diagnosis and management of acute deep vein thrombosis: A joint consensus document from the European Society of Cardiology working groups of aorta and peripheral vascular diseases and pulmonary circulation and right ventricular function. Eur. Hear. J. 2017, 39, 4208–4218.
- Kahn, S.R. The post-thrombotic syndrome. Hematology 2016, 2016, 413–418.
- Pulivarthi, S.; Gurram, M.e.K. Effectiveness of D-dimer as a screening test for venous thromboembolism: An update. North Am. J. Med. Sci. 2014, 6, 491–499.
- Righini, M.; Perrier, A.; De Moerloose, P.; Bounameaux, H. D-Dimer for venous thromboembolism diagnosis: 20 years later. J. Thromb. Haemost. 2008, 6, 1059–1071.
- Di Nisio, M.; Squizzato, A.; Rutjes, A.W.S.; Büller, H.R.; Zwinderman, A.H.; Bossuyt, P.M.M. Diagnostic accuracy of D-dimer test for exclusion of venous thromboembolism: A systematic review. J. Thromb. Haemost. 2007, 5, 296–304.
- Verhovsek, M.; Douketis, J.D.; Yi, Q.; Shrivastava, S.; Tait, R.C.; Baglin, T.; Poli, D.; Lim, W. Systematic Review: D-Dimer to Predict Recurrent Disease after Stopping Anticoagulant Therapy for Unprovoked Venous Thromboembolism. Ann. Intern. Med. 2008, 149, 481–490.
- Legnani, C.; Cosmi, B.; Guazzaloca, G.; Pancani, C.; Coccheri, S.; Palareti, G. Risk of Venous Thromboembolism Recurrence: High Negative Predictive Value of D-dimer Performed after Oral Anticoagulation Is Stopped. Thromb. Haemost. 2002, 87, 7–12.
- Eichinger, S.; Hron, G.; Kollars, M.; A Kyrle, P. Prediction of Recurrent Venous Thromboembolism by Endogenous Thrombin Potential and D-Dimer. Clin. Chem. 2008, 54, 2042–2048.
- van Veen, J.J.; Gatt, A.; Makris, M. Thrombin generation testing in routine clinical practice: Are we there yet? Br. J. Haematol. 2008, 142, 889–903.
- Brummel-Ziedins, K.E.; Vossen, C.Y.; Butenas, S.; Mann, K.G.; Rosendaal, F.R. Thrombin generation profiles in deep venous thrombosis. J. Thromb. Haemost. 2005, 3, 2497–2505.
- Lutsey, P.L.; Zakai, N.A. Epidemiology and prevention of venous thromboembolism. Nat. Rev. Cardiol. 2023, 20, 248–262.
- Haas, F.J.L.M.; Schutgens, R.E.G.; Kluft, C.; Biesma, D.H. A thrombin generation assay may reduce the need for compression ultrasonography for the exclusion of deep venous thrombosis in the elderly. Scand. J. Clin. Lab. Investig. 2011, 71, 12–18.
- McEver, R.P. Adhesive interactions of leukocytes, platelets, and the vessel wall during hemostasis and inflammation. Thromb. Haemost. 2001, 86.
- Ley, K. The role of selectins in inflammation and disease. Trends Mol. Med. 2003, 9, 263–268.
- Geng, J.-G.; Bevilacquat, M.P.; Moore, K.L.; Mclntyre, T.M.; Prescott, S.M.; Kim, J.M.; Bliss, G.A.; Zimmerman, G.A.; McEver, R.P. Rapid neutrophil adhesion to activated endothelium mediated by GMP-140. Nature 1990, 343, 757–760.
- Moore, K.L.; Stults, N.L.; Diaz, S.; Smith, D.F.; Cummings, R.D.; Varki, A.; McEver, R.P. Identification of a specific glycoprotein ligand for P-selectin (CD62) on myeloid cells. J. Cell Biol. 1992, 118, 445–456.
- Furie, B.; Furie, B.C. Role of platelet P-selectin and microparticle PSGL-1 in thrombus formation. Trends Mol. Med. 2004, 10, 171–178.
- Théorêt, J.-F.; Yacoub, D.; Hachem, A.; Gillis, M.-A.; Merhi, Y. P-selectin ligation induces platelet activation and enhances microaggregate and thrombus formation. Thromb. Res. 2011, 128, 243–250.
- Palabrica, T.; Lobb, R.; Furie, B.C.; Aronovitz, M.; Benjamin, C.; Hsu, Y.-M.; Sajer, S.A.; Furie, B. Leukocyte accumulation promoting fibrin deposition is mediated in vivo by P-selectin on adherent platelets. Nature 1992, 359, 848–851.
- Miszti-Blasius, K.; Debreceni, I.B.; Felszeghy, S.; Dezső, B.; Kappelmayer, J. Lack of P-selectin glycoprotein ligand-1 protects mice from thrombosis after collagen/epinephrine challenge. Thromb. Res. 2011, 127, 228–234.
- Vandy, F.C.; Stabler, C.; Eliassen, A.M.; Hawley, A.E.; Guire, K.E.; Myers, D.D.; Henke, P.K.; Wakefield, T.W. Soluble P-selectin for the diagnosis of lower extremity deep venous thrombosis. J. Vasc. Surg. Venous Lymphat. Disord. 2013, 1, 117–125.
- Hrachovinová, I.; Cambien, B.; Hafezi-Moghadam, A.; Kappelmayer, J.; Camphausen, R.T.; Widom, A.; Xia, L.; Kazazian, H.H., Jr.; Schaub, R.G.; McEver, R.P.; et al. Interaction of P-selectin and PSGL-1 generates microparticles that correct hemostasis in a mouse model of hemophilia A. Nat. Med. 2003, 9, 1020–1025.
- Rauch, U.; Bonderman, D.; Bohrmann, B.; Badimon, J.J.; Himber, J.; A Riederer, M.; Nemerson, Y. Transfer of tissue factor from leukocytes to platelets is mediated by CD15 and tissue factor. Blood 2000, 96, 170–175.
- Osnes, L.; Westvik, A.B.; Joø, G.-B.; Okkenhaug, C.; Kierulf, P. Inhibition of IL-1 induced tissue factor (TF) synthesis and procoagulant activity (PCA) in purified human monocytes by IL-4, IL-10 and IL-13. Cytokine 1996, 8, 822–827.
- Lutsey, P.L.; Astor, B.C.; Cushman, M.; Folsom, A.R. C-reactive protein and venous thromboembolism. Thromb. Haemost. 2009, 102, 615–619.
- Zacho, J.; Tybjærg-Hansen, A.; Nordestgaard, B.G. C-Reactive Protein and Risk of Venous Thromboembolism in the General Population. Arter. Thromb. Vasc. Biol. 2010, 30, 1672–1678.
- Viel, K.R.; Machiah, D.K.; Warren, D.M.; Khachidze, M.; Buil, A.; Fernstrom, K.; Souto, J.C.; Peralta, J.M.; Smith, T.; Blangero, J.; et al. A sequence variation scan of the coagulation factor VIII (FVIII) structural gene and associations with plasma FVIII activity levels. Blood 2007, 109, 3713–3724.
- Beckers, M.; Ruven, H.; Haas, F.; Doevendans, P.; Cate, H.T.; Prins, M.; Biesma, D. Single nucleotide polymorphisms in inflammation-related genes are associated with venous thromboembolism. Eur. J. Intern. Med. 2010, 21, 289–292.
- Vormittag, R.; Hsieh, K.; Kaider, A.; Minar, E.; Bialonczyk, C.; Hirschl, M.; Mannhalter, C.; Pabinger, I. Interleukin-6 and interleukin-6 promoter polymorphism (−174) G>C in patients with spontaneous venous thromboembolism. Thromb. Haemost. 2006, 95, 802–806.
- Christiansen, S.C.; Næss, I.A.; Cannegieter, S.C.; Hammerstrøm, J.; Rosendaal, F.R.; Reitsma, P.H. Inflammatory Cytokines as Risk Factors for a First Venous Thrombosis: A Prospective Population-Based Study. PLoS Med. 2006, 3, e334.
- Downing, L.J.; Strieter, R.M.; Kadell, A.M.; Wilke, C.A.; Austin, J.C.; Hare, B.D.; Burdick, M.D.; Greenfield, L.J.; Wakefield, T.W. IL-10 regulates thrombus-induced vein wall inflammation and thrombosis. J. Immunol. 1998, 161, 1471–1476.
- Myers, D.D.; Hawley, A.E.; Longo, C.; Henke, P.K.; Guire, K.E.; Schmaier, A.H.; Wakefield, T.W.; Rectenwald, J.E. D-dimer, P-selectin, and microparticles: Novel markers to predict deep venous thrombosis. Thromb. Haemost. 2005, 94, 1312–1317.
- Key, N.S.; Chantrathammachart, P.; Moody, P.W.; Chang, J.-Y. Membrane microparticles in VTE and cancer. Thromb. Res. 2010, 125 (Suppl. S2), S80–S83.
- Campello, E.; Spiezia, L.; Radu, C.M.; Bulato, C.; Castelli, M.; Gavasso, S.; Simioni, P. Endothelial, platelet, and tissue factor-bearing microparticles in cancer patients with and without venous thromboembolism. Thromb. Res. 2011, 127, 473–477.
- Chirinos, J.A.; Heresi, G.A.; Velasquez, H.; Jy, W.; Jimenez, J.J.; Ahn, E.; Horstman, L.L.; Soriano, A.O.; Zambrano, J.P.; Ahn, Y.S. Elevation of Endothelial Microparticles, Platelets, and Leukocyte Activation in Patients with Venous Thromboembolism. J. Am. Coll. Cardiol. 2005, 45, 1467–1471.
This entry is offline, you can click here to edit this entry!