Tyrosine phosphorylation constitutes an essential mechanism of signal transduction in eukaryotic cells. In the immune system, this process is widely represented, and in T cells, the first biochemical event that occurs after antigenic recognition seems to be the phosphorylation of the tyrosine residues included in the CD3 chains. For T cells to develop, differentiate and proliferate, proper intracellular signaling is required, and the negative regulation of such signaling pathways is crucial to prevent autoimmune diseases or severe immunodeficiencies. Although not understood in depth, it is becoming increasingly clear that multiple negative regulatory loops exist along the TCR-signaling cascade. The tyrosine kinases Lck and ZAP70, together with the transmembrane adaptor LAT, are essential players in the transduction of TCR early signals, and the elimination of any of them causes serious alterations in the TCR-signaling cascade.
- TCR
- CD3
- ITAMs
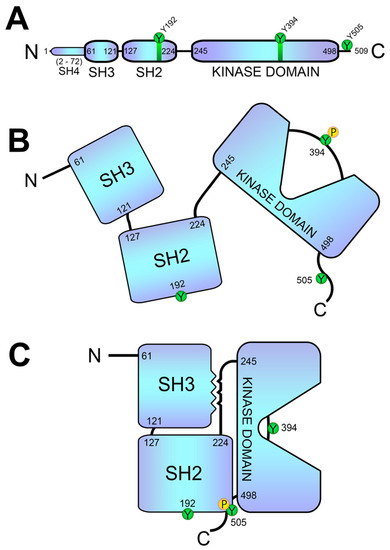
- Lck
- LAT adaptor
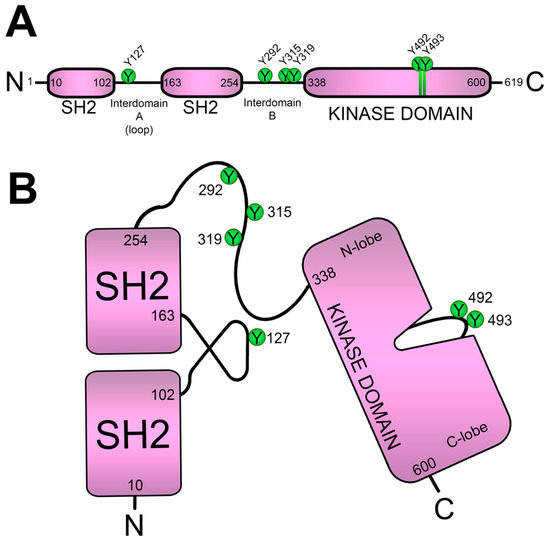
- ZAP70
1. Introduction
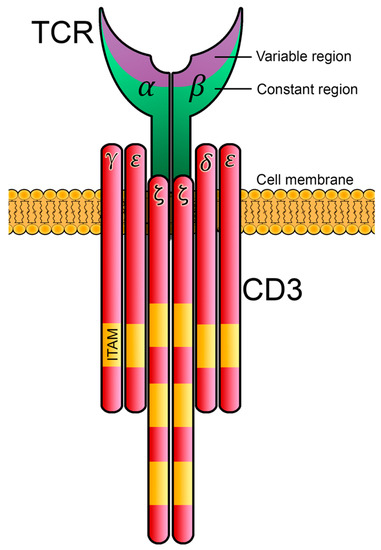
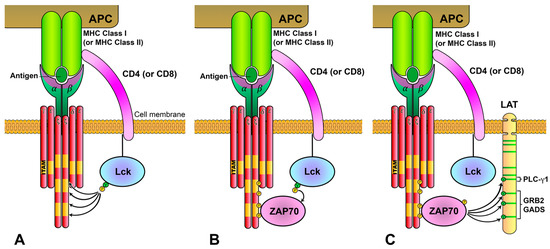
2. Initiation of Intracellular Signaling Associated with the TCR/CD3 Complex
3. Lck Goes into Action: The First Biochemical Events of the TCR-Signaling Cascade
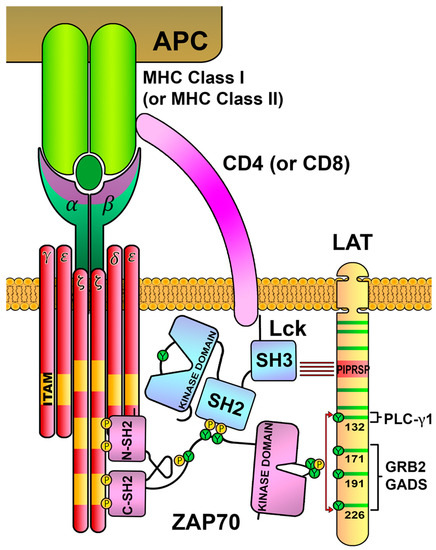
4. ZAP70 Is Recruited and Activated after ITAMs Phosphorylation
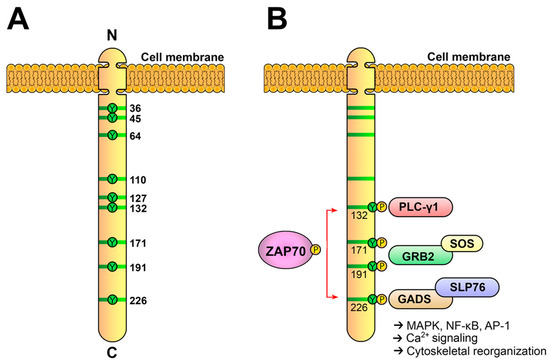
5. Activated ZAP70 Kinase Generates the LAT Signalosome
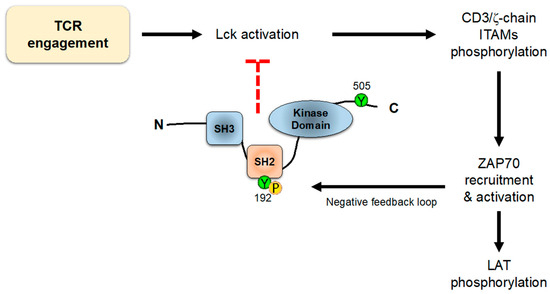
6. Regulation of Lck Activity
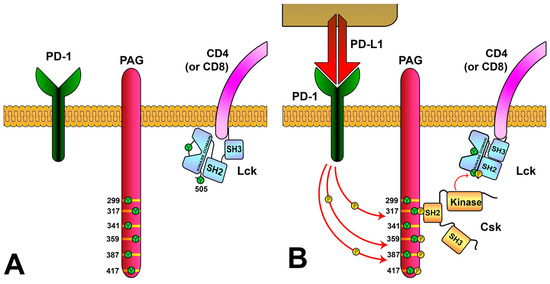
7. Keeping ZAP70 in Check
8. Negative-Feedback Loops Involving LAT
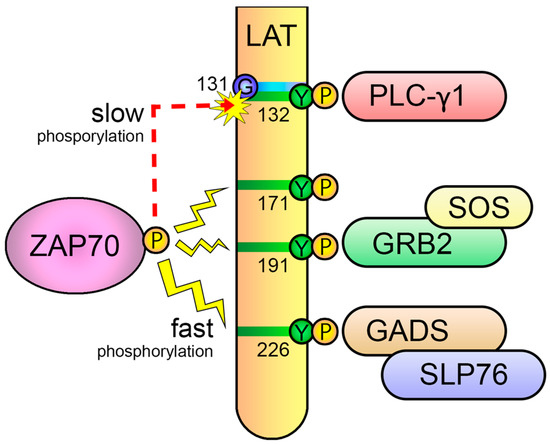
This entry is adapted from the peer-reviewed paper 10.3390/biology12091163
References
- Chakraborty, A.K.; Weiss, A. Insights into the initiation of TCR signaling. Nat. Immunol. 2014, 15, 798–807.
- Malissen, B.; Bongrand, P. Early T cell activation: Integrating biochemical, structural, and biophysical cues. Annu. Rev. Immunol. 2015, 33, 539–561.
- Chitadze, G.; Oberg, H.H.; Wesch, D.; Kabelitz, D. The Ambiguous Role of gammadelta T Lymphocytes in Antitumor Immunity. Trends Immunol. 2017, 38, 668–678.
- Alcover, A.; Alarcon, B.; Di Bartolo, V. Cell Biology of T Cell Receptor Expression and Regulation. Annu. Rev. Immunol. 2018, 36, 103–125.
- Schamel, W.W.; Alarcon, B. Organization of the resting TCR in nanoscale oligomers. Immunol. Rev. 2013, 251, 13–20.
- Reth, M. Antigen receptor tail clue. Nature 1989, 338, 383–384.
- Bezbradica, J.S.; Medzhitov, R. Role of ITAM signaling module in signal integration. Curr. Opin. Immunol. 2012, 24, 58–66.
- Palacios, E.H.; Weiss, A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene 2004, 23, 7990–8000.
- Ventimiglia, L.N.; Alonso, M.A. The role of membrane rafts in Lck transport, regulation and signalling in T-cells. Biochem. J. 2013, 454, 169–179.
- Malissen, B.; Aguado, E.; Malissen, M. Role of the LAT adaptor in T-cell development and Th2 differentiation. Adv. Immunol. 2005, 87, 1–25.
- Deng, L.; Luo, M.; Velikovsky, A.; Mariuzza, R.A. Structural insights into the evolution of the adaptive immune system. Annu. Rev. Biophys. 2013, 42, 191–215.
- Brameshuber, M.; Kellner, F.; Rossboth, B.K.; Ta, H.; Alge, K.; Sevcsik, E.; Gohring, J.; Axmann, M.; Baumgart, F.; Gascoigne, N.R.J.; et al. Monomeric TCRs drive T cell antigen recognition. Nat. Immunol. 2018, 19, 487–496.
- Cheng, Y.; Grigorieff, N.; Penczek, P.A.; Walz, T. A primer to single-particle cryo-electron microscopy. Cell 2015, 161, 438–449.
- Dong, D.; Zheng, L.; Lin, J.; Zhang, B.; Zhu, Y.; Li, N.; Xie, S.; Wang, Y.; Gao, N.; Huang, Z. Structural basis of assembly of the human T cell receptor-CD3 complex. Nature 2019, 573, 546–552.
- Hayes, S.M.; Love, P.E. Distinct structure and signaling potential of the gamma delta TCR complex. Immunity 2002, 16, 827–838.
- Siegers, G.M.; Swamy, M.; Fernandez-Malave, E.; Minguet, S.; Rathmann, S.; Guardo, A.C.; Perez-Flores, V.; Regueiro, J.R.; Alarcon, B.; Fisch, P.; et al. Different composition of the human and the mouse gammadelta T cell receptor explains different phenotypes of CD3gamma and CD3delta immunodeficiencies. J. Exp. Med. 2007, 204, 2537–2544.
- Champagne, E. gammadelta T cell receptor ligands and modes of antigen recognition. Arch. Immunol. Ther. Exp. (Warsz) 2011, 59, 117–137.
- Gil, D.; Schamel, W.W.; Montoya, M.; Sanchez-Madrid, F.; Alarcon, B. Recruitment of Nck by CD3 epsilon reveals a ligand-induced conformational change essential for T cell receptor signaling and synapse formation. Cell 2002, 109, 901–912.
- Szymczak, A.L.; Workman, C.J.; Gil, D.; Dilioglou, S.; Vignali, K.M.; Palmer, E.; Vignali, D.A. The CD3epsilon proline-rich sequence, and its interaction with Nck, is not required for T cell development and function. J. Immunol. 2005, 175, 270–275.
- Mingueneau, M.; Sansoni, A.; Gregoire, C.; Roncagalli, R.; Aguado, E.; Weiss, A.; Malissen, M.; Malissen, B. The proline-rich sequence of CD3epsilon controls T cell antigen receptor expression on and signaling potency in preselection CD4+CD8+ thymocytes. Nat. Immunol. 2008, 9, 522–532.
- Gil, D.; Schrum, A.G.; Alarcon, B.; Palmer, E. T cell receptor engagement by peptide-MHC ligands induces a conformational change in the CD3 complex of thymocytes. J. Exp. Med. 2005, 201, 517–522.
- Risueno, R.M.; van Santen, H.M.; Alarcon, B. A conformational change senses the strength of T cell receptor-ligand interaction during thymic selection. Proc. Natl. Acad. Sci. USA 2006, 103, 9625–9630.
- Risueno, R.M.; Gil, D.; Fernandez, E.; Sanchez-Madrid, F.; Alarcon, B. Ligand-induced conformational change in the T-cell receptor associated with productive immune synapses. Blood 2005, 106, 601–608.
- van der Merwe, P.A.; Dushek, O. Mechanisms for T cell receptor triggering. Nat. Rev. Immunol. 2011, 11, 47–55.
- Xu, C.; Gagnon, E.; Call, M.E.; Schnell, J.R.; Schwieters, C.D.; Carman, C.V.; Chou, J.J.; Wucherpfennig, K.W. Regulation of T cell receptor activation by dynamic membrane binding of the CD3epsilon cytoplasmic tyrosine-based motif. Cell 2008, 135, 702–713.
- Fernandes, R.A.; Yu, C.; Carmo, A.M.; Evans, E.J.; van der Merwe, P.A.; Davis, S.J. What controls T cell receptor phosphorylation? Cell 2010, 142, 668–669.
- Shi, X.; Bi, Y.; Yang, W.; Guo, X.; Jiang, Y.; Wan, C.; Li, L.; Bai, Y.; Guo, J.; Wang, Y.; et al. Ca2+ regulates T-cell receptor activation by modulating the charge property of lipids. Nature 2013, 493, 111–115.
- Lee, M.S.; Glassman, C.R.; Deshpande, N.R.; Badgandi, H.B.; Parrish, H.L.; Uttamapinant, C.; Stawski, P.S.; Ting, A.Y.; Kuhns, M.S. A Mechanical Switch Couples T Cell Receptor Triggering to the Cytoplasmic Juxtamembrane Regions of CD3zetazeta. Immunity 2015, 43, 227–239.
- Lanz, A.L.; Masi, G.; Porciello, N.; Cohnen, A.; Cipria, D.; Prakaash, D.; Balint, S.; Raggiaschi, R.; Galgano, D.; Cole, D.K.; et al. Allosteric activation of T cell antigen receptor signaling by quaternary structure relaxation. Cell Rep. 2021, 36, 109375.
- Susac, L.; Vuong, M.T.; Thomas, C.; von Bulow, S.; O’Brien-Ball, C.; Santos, A.M.; Fernandes, R.A.; Hummer, G.; Tampe, R.; Davis, S.J. Structure of a fully assembled tumor-specific T cell receptor ligated by pMHC. Cell 2022, 185, 3201–3213.
- Samelson, L.E.; Davidson, W.F.; Morse, H.C., 3rd; Klausner, R.D. Abnormal tyrosine phosphorylation on T-cell receptor in lymphoproliferative disorders. Nature 1986, 324, 674–676.
- Straus, D.B.; Weiss, A. Genetic evidence for the involvement of the lck tyrosine kinase in signal transduction through the T cell antigen receptor. Cell 1992, 70, 585–593.
- Marth, J.D.; Peet, R.; Krebs, E.G.; Perlmutter, R.M. A lymphocyte-specific protein-tyrosine kinase gene is rearranged and overexpressed in the murine T cell lymphoma LSTRA. Cell 1985, 43, 393–404.
- Marth, J.D.; Cooper, J.A.; King, C.S.; Ziegler, S.F.; Tinker, D.A.; Overell, R.W.; Krebs, E.G.; Perlmutter, R.M. Neoplastic transformation induced by an activated lymphocyte-specific protein tyrosine kinase (pp56lck). Mol. Cell Biol. 1988, 8, 540–550.
- Molina, T.J.; Kishihara, K.; Siderovski, D.P.; van Ewijk, W.; Narendran, A.; Timms, E.; Wakeham, A.; Paige, C.J.; Hartmann, K.U.; Veillette, A.; et al. Profound block in thymocyte development in mice lacking p56lck. Nature 1992, 357, 161–164.
- Rudd, C.E. How the Discovery of the CD4/CD8-p56(lck) Complexes Changed Immunology and Immunotherapy. Front. Cell Dev. Biol. 2021, 9, 626095.
- Yurchak, L.K.; Sefton, B.M. Palmitoylation of either Cys-3 or Cys-5 is required for the biological activity of the Lck tyrosine protein kinase. Mol. Cell Biol. 1995, 15, 6914–6922.
- Kabouridis, P.S.; Magee, A.I.; Ley, S.C. S-acylation of LCK protein tyrosine kinase is essential for its signalling function in T lymphocytes. EMBO J. 1997, 16, 4983–4998.
- Shaw, A.S.; Chalupny, J.; Whitney, J.A.; Hammond, C.; Amrein, K.E.; Kavathas, P.; Sefton, B.M.; Rose, J.K. Short related sequences in the cytoplasmic domains of CD4 and CD8 mediate binding to the amino-terminal domain of the p56lck tyrosine protein kinase. Mol. Cell Biol. 1990, 10, 1853–1862.
- Philipsen, L.; Reddycherla, A.V.; Hartig, R.; Gumz, J.; Kastle, M.; Kritikos, A.; Poltorak, M.P.; Prokazov, Y.; Turbin, E.; Weber, A.; et al. De novo phosphorylation and conformational opening of the tyrosine kinase Lck act in concert to initiate T cell receptor signaling. Sci. Signal 2017, 10, eaaf4736.
- Xu, R.X.; Word, J.M.; Davis, D.G.; Rink, M.J.; Willard, D.H., Jr.; Gampe, R.T., Jr. Solution structure of the human pp60c-src SH2 domain complexed with a phosphorylated tyrosine pentapeptide. Biochemistry 1995, 34, 2107–2121.
- Hui, E.; Vale, R.D. In vitro membrane reconstitution of the T-cell receptor proximal signaling network. Nat. Struct. Mol. Biol. 2014, 21, 133–142.
- Mustelin, T.; Coggeshall, K.M.; Altman, A. Rapid activation of the T-cell tyrosine protein kinase pp56lck by the CD45 phosphotyrosine phosphatase. Proc. Natl. Acad. Sci. USA 1989, 86, 6302–6306.
- Ostergaard, H.L.; Shackelford, D.A.; Hurley, T.R.; Johnson, P.; Hyman, R.; Sefton, B.M.; Trowbridge, I.S. Expression of CD45 alters phosphorylation of the lck-encoded tyrosine protein kinase in murine lymphoma T-cell lines. Proc. Natl. Acad. Sci. USA 1989, 86, 8959–8963.
- Sieh, M.; Bolen, J.B.; Weiss, A. CD45 specifically modulates binding of Lck to a phosphopeptide encompassing the negative regulatory tyrosine of Lck. EMBO J. 1993, 12, 315–321.
- Chan, A.C.; Irving, B.A.; Fraser, J.D.; Weiss, A. The zeta chain is associated with a tyrosine kinase and upon T-cell antigen receptor stimulation associates with ZAP-70, a 70-kDa tyrosine phosphoprotein. Proc. Natl. Acad. Sci. USA 1991, 88, 9166–9170.
- Chan, A.C.; Iwashima, M.; Turck, C.W.; Weiss, A. ZAP-70: A 70 kd protein-tyrosine kinase that associates with the TCR zeta chain. Cell 1992, 71, 649–662.
- Wange, R.L.; Malek, S.N.; Desiderio, S.; Samelson, L.E. Tandem SH2 domains of ZAP-70 bind to T cell antigen receptor zeta and CD3 epsilon from activated Jurkat T cells. J. Biol. Chem. 1993, 268, 19797–19801.
- Deindl, S.; Kadlecek, T.A.; Brdicka, T.; Cao, X.; Weiss, A.; Kuriyan, J. Structural basis for the inhibition of tyrosine kinase activity of ZAP-70. Cell 2007, 129, 735–746.
- Mege, D.; Di Bartolo, V.; Germain, V.; Tuosto, L.; Michel, F.; Acuto, O. Mutation of tyrosines 492/493 in the kinase domain of ZAP-70 affects multiple T-cell receptor signaling pathways. J. Biol. Chem. 1996, 271, 32644–32652.
- Bubeck Wardenburg, J.; Fu, C.; Jackman, J.K.; Flotow, H.; Wilkinson, S.E.; Williams, D.H.; Johnson, R.; Kong, G.; Chan, A.C.; Findell, P.R. Phosphorylation of SLP-76 by the ZAP-70 protein-tyrosine kinase is required for T-cell receptor function. J. Biol. Chem. 1996, 271, 19641–19644.
- Zhang, W.; Sloan-Lancaster, J.; Kitchen, J.; Trible, R.P.; Samelson, L.E. LAT: The ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell 1998, 92, 83–92.
- Arpaia, E.; Shahar, M.; Dadi, H.; Cohen, A.; Roifman, C.M. Defective T cell receptor signaling and CD8+ thymic selection in humans lacking zap-70 kinase. Cell 1994, 76, 947–958.
- Chan, A.C.; Kadlecek, T.A.; Elder, M.E.; Filipovich, A.H.; Kuo, W.L.; Iwashima, M.; Parslow, T.G.; Weiss, A. ZAP-70 deficiency in an autosomal recessive form of severe combined immunodeficiency. Science 1994, 264, 1599–1601.
- Elder, M.E.; Lin, D.; Clever, J.; Chan, A.C.; Hope, T.J.; Weiss, A.; Parslow, T.G. Human severe combined immunodeficiency due to a defect in ZAP-70, a T cell tyrosine kinase. Science 1994, 264, 1596–1599.
- Negishi, I.; Motoyama, N.; Nakayama, K.; Nakayama, K.; Senju, S.; Hatakeyama, S.; Zhang, Q.; Chan, A.C.; Loh, D.Y. Essential role for ZAP-70 in both positive and negative selection of thymocytes. Nature 1995, 376, 435–438.
- Au-Yeung, B.B.; Shah, N.H.; Shen, L.; Weiss, A. ZAP-70 in Signaling, Biology, and Disease. Annu. Rev. Immunol. 2018, 36, 127–156.
- Rassenti, L.Z.; Huynh, L.; Toy, T.L.; Chen, L.; Keating, M.J.; Gribben, J.G.; Neuberg, D.S.; Flinn, I.W.; Rai, K.R.; Byrd, J.C.; et al. ZAP-70 compared with immunoglobulin heavy-chain gene mutation status as a predictor of disease progression in chronic lymphocytic leukemia. N. Engl. J. Med. 2004, 351, 893–901.
- Chen, L.; Widhopf, G.; Huynh, L.; Rassenti, L.; Rai, K.R.; Weiss, A.; Kipps, T.J. Expression of ZAP-70 is associated with increased B-cell receptor signaling in chronic lymphocytic leukemia. Blood 2002, 100, 4609–4614.
- Gobessi, S.; Laurenti, L.; Longo, P.G.; Sica, S.; Leone, G.; Efremov, D.G. ZAP-70 enhances B-cell-receptor signaling despite absent or inefficient tyrosine kinase activation in chronic lymphocytic leukemia and lymphoma B cells. Blood 2007, 109, 2032–2039.
- Chen, L.; Huynh, L.; Apgar, J.; Tang, L.; Rassenti, L.; Weiss, A.; Kipps, T.J. ZAP-70 enhances IgM signaling independent of its kinase activity in chronic lymphocytic leukemia. Blood 2008, 111, 2685–2692.
- Chen, J.; Sathiaseelan, V.; Moore, A.; Tan, S.; Chilamakuri, C.S.R.; Roamio Franklin, V.N.; Shahsavari, A.; Jakwerth, C.A.; Hake, S.B.; Warren, A.J.; et al. ZAP-70 constitutively regulates gene expression and protein synthesis in chronic lymphocytic leukemia. Blood 2021, 137, 3629–3640.
- Au-Yeung, B.B.; Levin, S.E.; Zhang, C.; Hsu, L.Y.; Cheng, D.A.; Killeen, N.; Shokat, K.M.; Weiss, A. A genetically selective inhibitor demonstrates a function for the kinase Zap70 in regulatory T cells independent of its catalytic activity. Nat. Immunol. 2010, 11, 1085–1092.
- Jenkins, M.R.; Stinchcombe, J.C.; Au-Yeung, B.B.; Asano, Y.; Ritter, A.T.; Weiss, A.; Griffiths, G.M. Distinct structural and catalytic roles for Zap70 in formation of the immunological synapse in CTL. Elife 2014, 3, e01310.
- Aguado, E.; Martinez-Florensa, M.; Aparicio, P. Activation of T lymphocytes and the role of the adapter LAT. Transpl. Immunol. 2006, 17, 23–26.
- June, C.H.; Fletcher, M.C.; Ledbetter, J.A.; Samelson, L.E. Increases in tyrosine phosphorylation are detectable before phospholipase C activation after T cell receptor stimulation. J. Immunol. 1990, 144, 1591–1599.
- Nunes, J.A.; Truneh, A.; Olive, D.; Cantrell, D.A. Signal transduction by CD28 costimulatory receptor on T cells. B7-1 and B7-2 regulation of tyrosine kinase adaptor molecules. J. Biol. Chem. 1996, 271, 1591–1598.
- Finco, T.S.; Kadlecek, T.; Zhang, W.; Samelson, L.E.; Weiss, A. LAT is required for TCR-mediated activation of PLCgamma1 and the Ras pathway. Immunity 1998, 9, 617–626.
- Zhang, W.; Trible, R.P.; Zhu, M.; Liu, S.K.; McGlade, C.J.; Samelson, L.E. Association of Grb2, Gads, and phospholipase C-gamma 1 with phosphorylated LAT tyrosine residues. Effect of LAT tyrosine mutations on T cell angigen receptor-mediated signaling. J. Biol. Chem. 2000, 275, 23355–23361.
- Lin, J.; Weiss, A. Identification of the minimal tyrosine residues required for linker for activation of T cell function. J. Biol. Chem. 2001, 276, 29588–29595.
- Zhang, W.; Sommers, C.L.; Burshtyn, D.N.; Stebbins, C.C.; DeJarnette, J.B.; Trible, R.P.; Grinberg, A.; Tsay, H.C.; Jacobs, H.M.; Kessler, C.M.; et al. Essential role of LAT in T cell development. Immunity 1999, 10, 323–332.
- Chiesa, S.; Mingueneau, M.; Fuseri, N.; Malissen, B.; Raulet, D.H.; Malissen, M.; Vivier, E.; Tomasello, E. Multiplicity and plasticity of natural killer cell signaling pathways. Blood 2006, 107, 2364–2372.
- Sommers, C.L.; Menon, R.K.; Grinberg, A.; Zhang, W.; Samelson, L.E.; Love, P.E. Knock-in mutation of the distal four tyrosines of linker for activation of T cells blocks murine T cell development. J. Exp. Med. 2001, 194, 135–142.
- Zhu, M.; Janssen, E.; Zhang, W. Minimal requirement of tyrosine residues of linker for activation of T cells in TCR signaling and thymocyte development. J. Immunol. 2003, 170, 325–333.
- Aguado, E.; Richelme, S.; Nunez-Cruz, S.; Miazek, A.; Mura, A.M.; Richelme, M.; Guo, X.J.; Sainty, D.; He, H.T.; Malissen, B.; et al. Induction of T helper type 2 immunity by a point mutation in the LAT adaptor. Science 2002, 296, 2036–2040.
- Sommers, C.L.; Park, C.S.; Lee, J.; Feng, C.; Fuller, C.L.; Grinberg, A.; Hildebrand, J.A.; Lacana, E.; Menon, R.K.; Shores, E.W.; et al. A LAT mutation that inhibits T cell development yet induces lymphoproliferation. Science 2002, 296, 2040–2043.
- Songyang, Z.; Shoelson, S.E.; Chaudhuri, M.; Gish, G.; Pawson, T.; Haser, W.G.; King, F.; Roberts, T.; Ratnofsky, S.; Lechleider, R.J.; et al. SH2 domains recognize specific phosphopeptide sequences. Cell 1993, 72, 767–778.
- Paz, P.E.; Wang, S.; Clarke, H.; Lu, X.; Stokoe, D.; Abo, A. Mapping the Zap-70 phosphorylation sites on LAT (linker for activation of T cells) required for recruitment and activation of signalling proteins in T cells. Biochem. J. 2001, 356, 461–471.
- Su, X.; Ditlev, J.A.; Hui, E.; Xing, W.; Banjade, S.; Okrut, J.; King, D.S.; Taunton, J.; Rosen, M.K.; Vale, R.D. Phase separation of signaling molecules promotes T cell receptor signal transduction. Science 2016, 352, 595–599.
- Ditlev, J.A.; Vega, A.R.; Koster, D.V.; Su, X.; Tani, T.; Lakoduk, A.M.; Vale, R.D.; Mayor, S.; Jaqaman, K.; Rosen, M.K. A composition-dependent molecular clutch between T cell signaling condensates and actin. Elife 2019, 8, e42695.
- Wang, H.Y.; Chan, S.H.; Dey, S.; Castello-Serrano, I.; Rosen, M.K.; Ditlev, J.A.; Levental, K.R.; Levental, I. Coupling of protein condensates to ordered lipid domains determines functional membrane organization. Sci. Adv. 2023, 9, eadf6205.
- Irles, C.; Symons, A.; Michel, F.; Bakker, T.R.; van der Merwe, P.A.; Acuto, O. CD45 ectodomain controls interaction with GEMs and Lck activity for optimal TCR signaling. Nat. Immunol. 2003, 4, 189–197.
- Paster, W.; Paar, C.; Eckerstorfer, P.; Jakober, A.; Drbal, K.; Schutz, G.J.; Sonnleitner, A.; Stockinger, H. Genetically encoded Forster resonance energy transfer sensors for the conformation of the Src family kinase Lck. J. Immunol. 2009, 182, 2160–2167.
- Nika, K.; Soldani, C.; Salek, M.; Paster, W.; Gray, A.; Etzensperger, R.; Fugger, L.; Polzella, P.; Cerundolo, V.; Dushek, O.; et al. Constitutively active Lck kinase in T cells drives antigen receptor signal transduction. Immunity 2010, 32, 766–777.
- Ballek, O.; Valecka, J.; Manning, J.; Filipp, D. The pool of preactivated Lck in the initiation of T-cell signaling: A critical re-evaluation of the Lck standby model. Immunol. Cell Biol. 2015, 93, 384–395.
- Schoenborn, J.R.; Tan, Y.X.; Zhang, C.; Shokat, K.M.; Weiss, A. Feedback circuits monitor and adjust basal Lck-dependent events in T cell receptor signaling. Sci. Signal 2011, 4, ra59.
- Imamoto, A.; Soriano, P. Disruption of the csk gene, encoding a negative regulator of Src family tyrosine kinases, leads to neural tube defects and embryonic lethality in mice. Cell 1993, 73, 1117–1124.
- Tan, Y.X.; Manz, B.N.; Freedman, T.S.; Zhang, C.; Shokat, K.M.; Weiss, A. Inhibition of the kinase Csk in thymocytes reveals a requirement for actin remodeling in the initiation of full TCR signaling. Nat. Immunol. 2014, 15, 186–194.
- Manz, B.N.; Tan, Y.X.; Courtney, A.H.; Rutaganira, F.; Palmer, E.; Shokat, K.M.; Weiss, A. Small molecule inhibition of Csk alters affinity recognition by T cells. Elife 2015, 4, e08088.
- Chang, V.T.; Fernandes, R.A.; Ganzinger, K.A.; Lee, S.F.; Siebold, C.; McColl, J.; Jonsson, P.; Palayret, M.; Harlos, K.; Coles, C.H.; et al. Initiation of T cell signaling by CD45 segregation at ‘close contacts’. Nat. Immunol. 2016, 17, 574–582.
- Brdicka, T.; Pavlistova, D.; Leo, A.; Bruyns, E.; Korinek, V.; Angelisova, P.; Scherer, J.; Shevchenko, A.; Hilgert, I.; Cerny, J.; et al. Phosphoprotein associated with glycosphingolipid-enriched microdomains (PAG), a novel ubiquitously expressed transmembrane adaptor protein, binds the protein tyrosine kinase csk and is involved in regulation of T cell activation. J. Exp. Med. 2000, 191, 1591–1604.
- Davidson, D.; Bakinowski, M.; Thomas, M.L.; Horejsi, V.; Veillette, A. Phosphorylation-dependent regulation of T-cell activation by PAG/Cbp, a lipid raft-associated transmembrane adaptor. Mol. Cell Biol. 2003, 23, 2017–2028.
- Davidson, D.; Zhong, M.C.; Pandolfi, P.P.; Bolland, S.; Xavier, R.J.; Seed, B.; Li, X.; Gu, H.; Veillette, A. The Csk-Associated Adaptor PAG Inhibits Effector T Cell Activation in Cooperation with Phosphatase PTPN22 and Dok Adaptors. Cell Rep. 2016, 17, 2776–2788.
- Strazza, M.; Azoulay-Alfaguter, I.; Peled, M.; Adam, K.; Mor, A. Transmembrane adaptor protein PAG is a mediator of PD-1 inhibitory signaling in human T cells. Commun. Biol. 2021, 4, 672.
- Courtney, A.H.; Amacher, J.F.; Kadlecek, T.A.; Mollenauer, M.N.; Au-Yeung, B.B.; Kuriyan, J.; Weiss, A. A Phosphosite within the SH2 Domain of Lck Regulates Its Activation by CD45. Mol. Cell 2017, 67, 498–511.e6.
- Kastle, M.; Merten, C.; Hartig, R.; Kaehne, T.; Liaunardy-Jopeace, A.; Woessner, N.M.; Schamel, W.W.; James, J.; Minguet, S.; Simeoni, L.; et al. Tyrosine 192 within the SH2 domain of the Src-protein tyrosine kinase p56(Lck) regulates T-cell activation independently of Lck/CD45 interactions. Cell Commun. Signal 2020, 18, 183.
- Kastle, M.; Merten, C.; Hartig, R.; Plaza-Sirvent, C.; Schmitz, I.; Bommhardt, U.; Schraven, B.; Simeoni, L. Y192 within the SH2 Domain of Lck Regulates TCR Signaling Downstream of PLC-gamma1 and Thymic Selection. Int. J. Mol. Sci. 2022, 23, 7271.
- Montixi, C.; Langlet, C.; Bernard, A.M.; Thimonier, J.; Dubois, C.; Wurbel, M.A.; Chauvin, J.P.; Pierres, M.; He, H.T. Engagement of T cell receptor triggers its recruitment to low-density detergent-insoluble membrane domains. EMBO J. 1998, 17, 5334–5348.
- Drevot, P.; Langlet, C.; Guo, X.J.; Bernard, A.M.; Colard, O.; Chauvin, J.P.; Lasserre, R.; He, H.T. TCR signal initiation machinery is pre-assembled and activated in a subset of membrane rafts. EMBO J. 2002, 21, 1899–1908.
- Urbancic, I.; Schiffelers, L.; Jenkins, E.; Gong, W.; Santos, A.M.; Schneider, F.; O’Brien-Ball, C.; Vuong, M.T.; Ashman, N.; Sezgin, E.; et al. Aggregation and mobility of membrane proteins interplay with local lipid order in the plasma membrane of T cells. FEBS Lett. 2021, 595, 2127–2146.
- Porciello, N.; Cipria, D.; Masi, G.; Lanz, A.L.; Milanetti, E.; Grottesi, A.; Howie, D.; Cobbold, S.P.; Schermelleh, L.; He, H.T.; et al. Role of the membrane anchor in the regulation of Lck activity. J. Biol. Chem. 2022, 298, 102663.
- Prakaash, D.; Fagnen, C.; Cook, G.P.; Acuto, O.; Kalli, A.C. Molecular dynamics simulations reveal membrane lipid interactions of the full-length lymphocyte specific kinase (Lck). Sci. Rep. 2022, 12, 21121.
- Liang, Y.; Ye, L. Bound to be perfect: Lck and T cell co-receptors. Nat. Immunol. 2023, 24, 5–7.
- Horkova, V.; Drobek, A.; Paprckova, D.; Niederlova, V.; Prasai, A.; Uleri, V.; Glatzova, D.; Kraller, M.; Cesnekova, M.; Janusova, S.; et al. Unique roles of co-receptor-bound LCK in helper and cytotoxic T cells. Nat. Immunol. 2023, 24, 174–185.
- Van Laethem, F.; Tikhonova, A.N.; Pobezinsky, L.A.; Tai, X.; Kimura, M.Y.; Le Saout, C.; Guinter, T.I.; Adams, A.; Sharrow, S.O.; Bernhardt, G.; et al. Lck availability during thymic selection determines the recognition specificity of the T cell repertoire. Cell 2013, 154, 1326–1341.
- Di Bartolo, V.; Mege, D.; Germain, V.; Pelosi, M.; Dufour, E.; Michel, F.; Magistrelli, G.; Isacchi, A.; Acuto, O. Tyrosine 319, a newly identified phosphorylation site of ZAP-70, plays a critical role in T cell antigen receptor signaling. J. Biol. Chem. 1999, 274, 6285–6294.
- Pelosi, M.; Di Bartolo, V.; Mounier, V.; Mege, D.; Pascussi, J.M.; Dufour, E.; Blondel, A.; Acuto, O. Tyrosine 319 in the interdomain B of ZAP-70 is a binding site for the Src homology 2 domain of Lck. J. Biol. Chem. 1999, 274, 14229–14237.
- Marquez, M.E.; Ellmeier, W.; Sanchez-Guajardo, V.; Freitas, A.A.; Acuto, O.; Di Bartolo, V. CD8 T cell sensory adaptation dependent on TCR avidity for self-antigens. J. Immunol. 2005, 175, 7388–7397.
- Brdicka, T.; Kadlecek, T.A.; Roose, J.P.; Pastuszak, A.W.; Weiss, A. Intramolecular regulatory switch in ZAP-70: Analogy with receptor tyrosine kinases. Mol. Cell Biol. 2005, 25, 4924–4933.
- Magnan, A.; Di Bartolo, V.; Mura, A.M.; Boyer, C.; Richelme, M.; Lin, Y.L.; Roure, A.; Gillet, A.; Arrieumerlou, C.; Acuto, O.; et al. T cell development and T cell responses in mice with mutations affecting tyrosines 292 or 315 of the ZAP-70 protein tyrosine kinase. J. Exp. Med. 2001, 194, 491–505.
- Gong, Q.; Jin, X.; Akk, A.M.; Foger, N.; White, M.; Gong, G.; Bubeck Wardenburg, J.; Chan, A.C. Requirement for tyrosine residues 315 and 319 within zeta chain-associated protein 70 for T cell development. J. Exp. Med. 2001, 194, 507–518.
- Davanture, S.; Leignadier, J.; Milani, P.; Soubeyran, P.; Malissen, B.; Malissen, M.; Schmitt-Verhulst, A.M.; Boyer, C. Selective defect in antigen-induced TCR internalization at the immune synapse of CD8 T cells bearing the ZAP-70(Y292F) mutation. J. Immunol. 2005, 175, 3140–3149.
- Hsu, L.Y.; Tan, Y.X.; Xiao, Z.; Malissen, M.; Weiss, A. A hypomorphic allele of ZAP-70 reveals a distinct thymic threshold for autoimmune disease versus autoimmune reactivity. J. Exp. Med. 2009, 206, 2527–2541.
- Deindl, S.; Kadlecek, T.A.; Cao, X.; Kuriyan, J.; Weiss, A. Stability of an autoinhibitory interface in the structure of the tyrosine kinase ZAP-70 impacts T cell receptor response. Proc. Natl. Acad. Sci. USA 2009, 106, 20699–20704.
- Yan, Q.; Barros, T.; Visperas, P.R.; Deindl, S.; Kadlecek, T.A.; Weiss, A.; Kuriyan, J. Structural basis for activation of ZAP-70 by phosphorylation of the SH2-kinase linker. Mol. Cell Biol. 2013, 33, 2188–2201.
- Hsu, L.Y.; Cheng, D.A.; Chen, Y.; Liang, H.E.; Weiss, A. Destabilizing the autoinhibitory conformation of Zap70 induces up-regulation of inhibitory receptors and T cell unresponsiveness. J. Exp. Med. 2017, 214, 833–849.
- Levin, S.E.; Zhang, C.; Kadlecek, T.A.; Shokat, K.M.; Weiss, A. Inhibition of ZAP-70 kinase activity via an analog-sensitive allele blocks T cell receptor and CD28 superagonist signaling. J. Biol. Chem. 2008, 283, 15419–15430.
- Goodfellow, H.S.; Frushicheva, M.P.; Ji, Q.; Cheng, D.A.; Kadlecek, T.A.; Cantor, A.J.; Kuriyan, J.; Chakraborty, A.K.; Salomon, A.; Weiss, A. The catalytic activity of the kinase ZAP-70 mediates basal signaling and negative feedback of the T cell receptor pathway. Sci. Signal 2015, 8, ra49.
- Xie, J.; Han, X.; Zhao, C.; Canonigo-Balancio, A.J.; Yates, J.R., 3rd; Li, Y.; Lillemeier, B.F.; Altman, A. Phosphotyrosine-dependent interaction between the kinases PKCtheta and Zap70 promotes proximal TCR signaling. Sci. Signal 2019, 12, eaar3349.
- Turul, T.; Tezcan, I.; Artac, H.; de Bruin-Versteeg, S.; Barendregt, B.H.; Reisli, I.; Sanal, O.; van Dongen, J.J.; van der Burg, M. Clinical heterogeneity can hamper the diagnosis of patients with ZAP70 deficiency. Eur. J. Pediatr. 2009, 168, 87–93.
- Schultz, A.; Schnurra, M.; El-Bizri, A.; Woessner, N.M.; Hartmann, S.; Hartig, R.; Minguet, S.; Schraven, B.; Simeoni, L. A Cysteine Residue within the Kinase Domain of Zap70 Regulates Lck Activity and Proximal TCR Signaling. Cells 2022, 11, 2723.
- Kabouridis, P.S. Selective interaction of LAT (linker of activated T cells) with the open-active form of Lck in lipid rafts reveals a new mechanism for the regulation of Lck in T cells. Biochem. J. 2003, 371, 907–915.
- Kabouridis, P.S.; Isenberg, D.A.; Jury, E.C. A negatively charged domain of LAT mediates its interaction with the active form of Lck. Mol. Membr. Biol. 2011, 28, 487–494.
- Arbulo-Echevarria, M.M.; Narbona-Sanchez, I.; Fernandez-Ponce, C.M.; Vico-Barranco, I.; Rueda-Ygueravide, M.D.; Dustin, M.L.; Miazek, A.; Duran-Ruiz, M.C.; Garcia-Cozar, F.; Aguado, E. A Stretch of Negatively Charged Amino Acids of Linker for Activation of T-Cell Adaptor Has a Dual Role in T-Cell Antigen Receptor Intracellular Signaling. Front. Immunol. 2018, 9, 115.
- Lo, W.L.; Shah, N.H.; Ahsan, N.; Horkova, V.; Stepanek, O.; Salomon, A.R.; Kuriyan, J.; Weiss, A. Lck promotes Zap70-dependent LAT phosphorylation by bridging Zap70 to LAT. Nat. Immunol. 2018, 19, 733–741.
- Strauss, G.; Lindquist, J.A.; Arhel, N.; Felder, E.; Karl, S.; Haas, T.L.; Fulda, S.; Walczak, H.; Kirchhoff, F.; Debatin, K.M. CD95 co-stimulation blocks activation of naive T cells by inhibiting T cell receptor signaling. J. Exp. Med. 2009, 206, 1379–1393.
- Berry, D.M.; Benn, S.J.; Cheng, A.M.; McGlade, C.J. Caspase-dependent cleavage of the hematopoietic specific adaptor protein Gads alters signalling from the T cell receptor. Oncogene 2001, 20, 1203–1211.
- Yankee, T.M.; Draves, K.E.; Ewings, M.K.; Clark, E.A.; Graves, J.D. CD95/Fas induces cleavage of the GrpL/Gads adaptor and desensitization of antigen receptor signaling. Proc. Natl. Acad. Sci. USA 2001, 98, 6789–6793.
- Arbulo-Echevarria, M.M.; Munoz-Miranda, J.P.; Caballero-Garcia, A.; Poveda-Diaz, J.L.; Fernandez-Ponce, C.; Duran-Ruiz, M.C.; Miazek, A.; Garcia-Cozar, F.; Aguado, E. Non-T cell activation linker (NTAL) proteolytic cleavage as a terminator of activatory intracellular signals. J. Leukoc. Biol. 2016, 100, 351–360.
- Bacchelli, C.; Moretti, F.A.; Carmo, M.; Adams, S.; Stanescu, H.C.; Pearce, K.; Madkaikar, M.; Gilmour, K.C.; Nicholas, A.K.; Woods, C.G.; et al. Mutations in linker for activation of T cells (LAT) lead to a novel form of severe combined immunodeficiency. J. Allergy Clin. Immunol. 2017, 139, 634–642.
- Keller, B.; Zaidman, I.; Yousefi, O.S.; Hershkovitz, D.; Stein, J.; Unger, S.; Schachtrup, K.; Sigvardsson, M.; Kuperman, A.A.; Shaag, A.; et al. Early onset combined immunodeficiency and autoimmunity in patients with loss-of-function mutation in LAT. J. Exp. Med. 2016, 213, 1185–1199.
- Courtney, A.H.; Lo, W.L.; Weiss, A. TCR Signaling: Mechanisms of Initiation and Propagation. Trends Biochem. Sci. 2018, 43, 108–123.
- Houtman, J.C.; Houghtling, R.A.; Barda-Saad, M.; Toda, Y.; Samelson, L.E. Early phosphorylation kinetics of proteins involved in proximal TCR-mediated signaling pathways. J. Immunol. 2005, 175, 2449–2458.
- Shah, N.H.; Wang, Q.; Yan, Q.; Karandur, D.; Kadlecek, T.A.; Fallahee, I.R.; Russ, W.P.; Ranganathan, R.; Weiss, A.; Kuriyan, J. An electrostatic selection mechanism controls sequential kinase signaling downstream of the T cell receptor. Elife 2016, 5, e20105.
- McKeithan, T.W. Kinetic proofreading in T-cell receptor signal transduction. Proc. Natl. Acad. Sci. USA 1995, 92, 5042–5046.
- Lo, W.L.; Shah, N.H.; Rubin, S.A.; Zhang, W.; Horkova, V.; Fallahee, I.R.; Stepanek, O.; Zon, L.I.; Kuriyan, J.; Weiss, A. Slow phosphorylation of a tyrosine residue in LAT optimizes T cell ligand discrimination. Nat. Immunol. 2019, 20, 1481–1493.
- Voisinne, G.; Locard-Paulet, M.; Froment, C.; Maturin, E.; Menoita, M.G.; Girard, L.; Mellado, V.; Burlet-Schiltz, O.; Malissen, B.; Gonzalez de Peredo, A.; et al. Kinetic proofreading through the multi-step activation of the ZAP70 kinase underlies early T cell ligand discrimination. Nat. Immunol. 2022, 23, 1355–1364.
- Arbulo-Echevarria, M.M.; Vico-Barranco, I.; Narbona-Sanchez, I.; Garcia-Cozar, F.; Miazek, A.; Aguado, E. Increased Protein Stability and Interleukin-2 Production of a LAT(G131D) Variant With Possible Implications for T Cell Anergy. Front. Cell Dev. Biol. 2020, 8, 561503.
- Arbulo-Echevarria, M.M.; Vico-Barranco, I.; Zhang, F.; Fernandez-Aguilar, L.M.; Chotomska, M.; Narbona-Sanchez, I.; Zhang, L.; Malissen, B.; Liang, Y.; Aguado, E. Mutation of the glycine residue preceding the sixth tyrosine of the LAT adaptor severely alters T cell development and activation. Front. Immunol. 2022, 13, 1054920.
- Lo, W.L.; Kuhlmann, M.; Rizzuto, G.; Ekiz, H.A.; Kolawole, E.M.; Revelo, M.P.; Andargachew, R.; Li, Z.; Tsai, Y.L.; Marson, A.; et al. A single-amino acid substitution in the adaptor LAT accelerates TCR proofreading kinetics and alters T-cell selection, maintenance and function. Nat. Immunol. 2023, 24, 676–689.