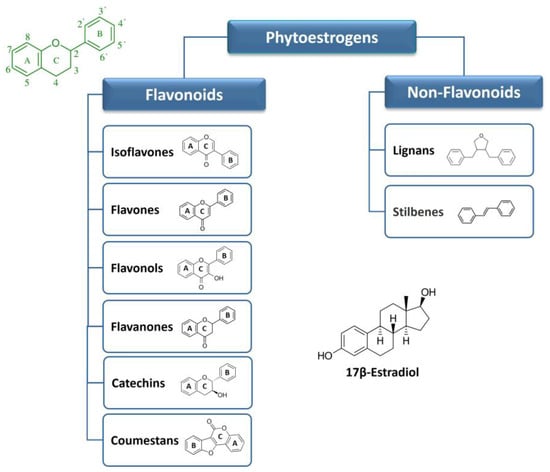
The actions of phytoestrogens via ERs can be mediated by genomic and/or non-genomic mechanisms, in a dose- and tissue-specific manner [
52,
56]. The ER-mediated genomic effects of phytoestrogens result in the regulation of target genes, which include anti-inflammatory, anti-apoptotic, metabolic, and mitochondrial genes, as well as an improvement in mitochondrial biogenesis and function, which leads to increased resistance to stress [
57,
58] (
Figure 2).
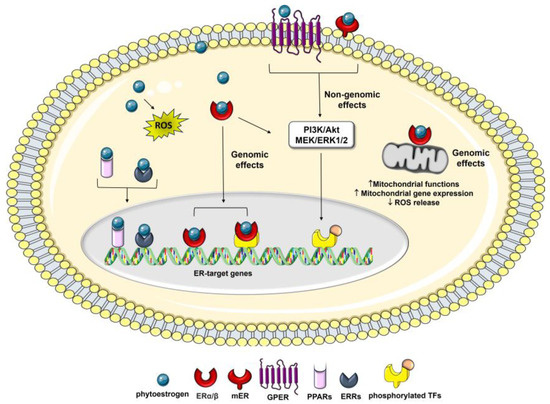
Figure 2. Intracellular mediators of the effects of phytoestrogens on cancer cells. Phytoestrogen can interact with the two types of ER, the intracellular ERα and Erβ, and membrane-associated mERs and GPER, activating downstream genomic and non-genomic effects which ultimately affect cell cancer phenotypes. The genomic pathway can involve ER interactions with other transcription factors (TFs), such as CREB, AP-1, Sp1, and NF-κB. Phytoestrogens can also act through ER-independent mechanisms which induce oxidative stress-mediated signaling by generating ROS, as well as interacting with other nuclear receptors, such as PPARs and ERRα/γ.
Phytoestrogens also modulate several therapeutically important oncogenic signaling pathways, including the epithelial–mesenchymal transition (EMT) and MAPK-associated pathways [
46,
59], and recruit transcription factors, such as response element binding protein (CREB), the activator protein 1 (AP-1), the stimulating protein 1 (Sp1), and the nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB), which are correlated with cell cycle regulation, angiogenesis, metastasis, and apoptosis [
60].
Besides nuclear events mediated by intracellular ER binding, many of these compounds also exert non-genomic effects through the activation of the membrane-associated ERs (mERs) and/or G-protein-coupled estrogen receptor 1 (GPER1/GPR30), which are involved in a diverse array of disorders, including cancer [
61,
62,
63]. Among the membrane-initiated non-genomic effects is the activation of signaling cascades, such as the mitogen-activated protein extracellular kinase/extracellular signal-regulated kinase (MEK/ERK) and PI3K pathways, which affect cancer cell apoptosis and proliferation [
10,
64].
In addition, they can exert estrogenic activity by cross-talk with many other pathways, including those related to membrane-associated growth factor receptors, such as the human epidermal growth factor receptor (EGFR/HER) and the insulin-like growth factor 1-receptor (IGF1R) [
65], as well as nuclear receptors, including [
66] peroxisome proliferator-activated receptors (PPARs) [
67] and estrogen-related receptor alpha/gamma (ERRα/γ) (
Figure 2) [
68]. Moreover, phytoestrogens can promote apoptosis and prevent the reproduction of malignant cells by blocking neo-angiogenesis, tyrosine-kinase, and topoisomerase proteins [
69]. Several studies have also reported that, in addition to the classical estrogen receptor signaling and the genomic and non-genomic effects mentioned above, some phytoestrogens, including genistein and resveratrol, exert their anticancer effects by the epigenetic mechanism, such as the modulation of the chromatin structure [
42,
70] and the regulation of different cancer-associated miRNAs [
71,
72], suggesting new therapeutic strategies for cancer.
Phytoestrogens are mostly known for their potent antioxidant activity, i.e., another biological activity that indirectly reduces the risk of various degenerative diseases linked to oxidative stress, including cancer [
73]. The chemical structures of these compounds consist of a long-conjugated system that includes phenolic groups. This structural arrangement confers them significant antioxidant properties [
8], which have been linked to their chemoprevention potential, particularly in Asian populations. It is worth noting that in these populations, there is a correlation between soy consumption and a reduced occurrence of estrogen-related cancers [
8,
24].
On the other hand, at high concentrations, phytoestrogens may have pro-oxidant effects and induce cell death. Flavonoids autoxidize in aqueous medium and may form highly reactive radicals in the presence of transition metals. This effect has been described for several compounds, including genistein [
74,
75] and resveratrol [
76], suggesting that a combination of phytoestrogens with anti-cancer treatments may render cancer cells more sensitive to treatment, in part by increasing the production of reactive oxygen species (ROS). However, given the high concentration of these compounds required for these activities, their impact on cancer onset and progression appears to be related to other cellular effects besides the modulation of oxidative stress [
77,
78].
Some phytoestrogens have also been shown to possess anti-inflammatory properties and modulate immune responses. They can inhibit the production of inflammatory mediators and reduce the expression of pro-inflammatory genes, contributing to their potential as anticancer agents [
8,
15].
As a whole, phytoestrogens exert a plethora of effects through multiple synergistic signaling pathways, which contribute to the outcome of phytoestrogen exposure on health and/or cancer cells. The specific effects of phytoestrogen exposure on cancer initiation, progression, and development may differ depending on the ERα/ERβ ratio in the affected tissue and the different selectivity and concentration of phytoestrogens [
48]. In this regard, the majority of reported findings indicate distinct effects at low and high concentrations of phytoestrogens, which may be attributed to the capacity of these molecules to interact with and modulate ERs, thereby influencing endocrine functions [
79]. Indeed, some studies have raised concerns about the potential adverse effects of soy products, particularly in high doses or when consumed by individuals with hormone-sensitive cancers [
10,
42]. Hence, it is crucial to gain a comprehensive understanding of how phytoestrogens interact with the ERs to fully evaluate their toxicologic and pharmacologic properties.
4. Molecular Basis of Osteosarcoma Pathogeneses
The difficulty in establishing an efficacious OS therapy is linked to the unclear specific markers for diagnosis and treatment. It is also due to the complexity of the OS genome, low incidence of this tumor, and significant biologic differences between OS subtypes. Nevertheless, the heterogeneity in the genotype of OS has translated into several expression profiles of macromolecular biomarkers, which are helpful in the clinic [
2,
80,
81,
82]. There are many genetic mutations observed in OS patients. The p53 and retinoblastoma (Rb) genes are well-known tumor-suppressor genes. Both germline and somatic mutations of the p53 and Rb genes have been proven to be involved in OS pathogenesis [
82,
83,
84]. Inherited cancer predisposition syndromes, such as Li–Fraumeni, hereditary retinoblastoma, Rothmund–Thomson, Bloom, or Werner syndrome, may also influence the high appearance of this kind of tumor in young patients [
83,
85,
86,
87,
88]. Among other genes mutated in more than 10% of OS cases, c-Myc plays a role in OS development and promotes cell invasion by activating MEK–ERK pathways. A high expression of c-Myc in OS tumors correlates with the formation of metastasis and poor prognosis [
89].
Several studies have consistently demonstrated that OS cells have the capacity to develop and secrete a range of growth factors that exert autocrine and paracrine effects. Vascular endothelial growth factor (VEGF), transforming growth factor (TGF), IGF-I and IGF-II, and connective tissue growth factor (CTGF) are deregulated in OS, which leads to tumor progression and growth in target cells [
82,
90,
91,
92]. Parathyroid hormone-related peptide (PTHrP) and its receptor have also been implicated in OS progression and metastasis development, with PTHrP conferring OS chemoresistance by blocking signaling via p53 [
93]. Epigenetic events have emerged as significant risk factors for OS, since the DNA methylation pattern of specific genes or gene regions and histone modifications may be involved in tumor development [
94]. In addition, a variety of studies have found abnormally expressed levels of micro-RNAs (miRs), which have the potential to become prognostic biomarkers of OS. Overexpression of this molecule results in proliferation, migration, and invasion of tumor cells [
68,
95]. Among the miRNAs deregulated in osteosarcoma are miR-421, miR-16, miR-200b, and miR-101 [
81,
96,
97].
OS is a highly metastatic tumor, and pulmonary metastases are the most common cause of death [
82,
98]. The ability of OS cells to metastasize has been found to be correlated with multiple processes and various cytophysiological changes, including changing the adhesion capabilities between cells and the extracellular matrix (ECM) and disrupting intercellular interactions [
99,
100]. Degradation of the ECM and components of the basement membrane caused by the concerted action of proteinases, such as matrix metalloproteinases (MMPs), cathepsins, and the plasminogen activator (PA), can play a critical role in OS invasion and metastasis [
100]. Moreover, in metastatic forms of OS, some specific genetic changes have been observed, which include upregulation of the Wnt/β-catenin and Src pathways, the neurogenic locus notch homolog protein 1 and 2 (Notch1/Notch2) receptors [
101,
102] together with the downregulation of the Fas/Fas ligand pathway (a cell death pathway), which increases the metastatic potential of human OS [
103,
104].
In both primary bone cancer and bone metastases, the bone remodeling process creates a favorable environment for tumor establishment and progression. Osteoblasts and osteoclasts are the primary regulators of bone metabolism [
105]. Specifically, osteoblasts secrete multiple components of ECM and MMPs in the OS niche, which are rich promoters of OS development. Moreover, osteoclasts play a pivotal role as bone-resorbing cells, and significant osteolysis exhibited in some OS cases can be directly attributed to the heightened activity of osteoclasts [
100].
It has been demonstrated that OS is a condition characterized by deregulation in the signaling triad, i.e., the receptor activator of nuclear factor kB Ligand (RANKL), its receptors RANK, and osteoprotegerin (OPG) [
106,
107]. In its canonical function, RANKL, which is secreted by osteoblasts, induces bone destruction by mature osteoclasts. In response, osteoblasts secrete the OPG–RANKL decoy receptor and in this way inhibit osteoclast differentiation and resultant bone resorption [
106,
108]. The RANKL/OPG ratio in the blood is increased in high-grade OS, leading to the establishment of a vicious cycle between pathological bone remodeling and OS growth [
108]. RANKL/RANK-signaling regulates OS cell migration and tissue-specific metastatic behavior in the lungs, but has no direct impact on OS-associated bone destruction and does not impact OS cell proliferation [
106,
109]. Thus, osteoclast pathways of differentiation, maturation, and activation constitute another compelling therapeutic target since the inhibition of bone resorption at the tumor–bone interface may lead to reduced local OS invasion [
106].
Among the possible mechanisms that contribute to OS development in the bone microenvironment are alterations in the osteogenic pathway, which lead to the differentiation of mesenchymal stem cells (MSCs) into mature osteoblasts [
81,
110]. Defects in osteogenic differentiation or exposure to new non-native stimuli, such as pro-inflammatory cytokines and pro-tumor agents, may cause an imbalance between cell differentiation and proliferation, thus contributing to a malignant phenotype. OS cells share more characteristics with undifferentiated osteo-progenitors than with differentiated osteoblasts, including a high proliferative capacity and resistance to apoptosis. Indeed, osteogenic regulators associated with mature osteoblast phenotypes, such as CTGF, RUNX2, alkaline phosphatase (ALP), osteopontin (OPN), and osteocalcin (OCN), are very lowly expressed in both primary OS tumors and OS cell lines [
111].
Although not well understood, some of the potential defects in the MSC differentiation cascade may include genetic and/or epigenetic changes in Wnt signaling, Rb, and p53. These alterations may lead to uncontrolled cell proliferation and disrupted differentiation, thus producing a tumorigenic phenotype [
81,
110]. Interestingly, treatments of human OS cells with therapeutic agents, such as peroxisome proliferator-activated receptor (PPAR) agonists [
111], growth factors (e.g., PTHrP) [
112], and SERMs [
113], enable terminal differentiation and subsequent tumor inhibition. Hence, a better understanding of the relationship between defects in osteogenic differentiation and tumor development is of fundamental importance for the treatment of OS and promoting differentiation offers a potential for disease control.