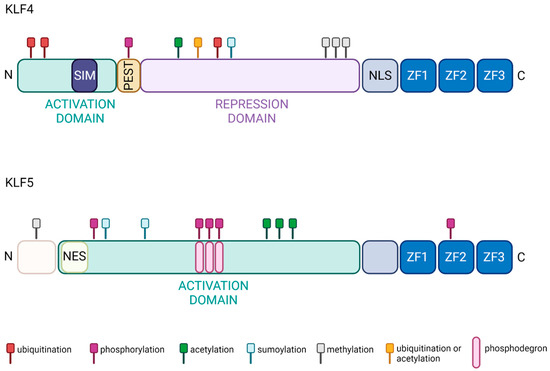
KLF4 is also called gut-enriched Krüppel-like factor (GKLF). KLF4 regulates transcription by modulating histone H4 acetylation at promoter sites [
27]. An overview of its biochemical properties, regulation, and physiological functions was reviewed by us elsewhere [
21]. KLF4 plays a significant role during gastrointestinal development and subsequent epithelial homeostasis. During murine fetal development, gastrointestinal KLF4 levels rise on embryonic day 13 and peak at day 17 [
59]. By birth, KLF4 levels in colonic cells are typically higher than in small intestine cells. KLF4 levels persist throughout the gastrointestinal tract during life and rise with increasing age throughout adulthood [
59]. Specifically, KLF4 is expressed in terminally differentiated epithelial cells at the mucosal villus border and reaches peak levels at terminal differentiation [
60,
61,
62,
63]. It is involved in goblet cell differentiation and maintenance and regulation of cell polarity [
61]. Conditional ablation of
Klf4 from the intestinal tract resulted in viable mice but with increased rates of epithelial proliferation and migration [
64]. Partial depletion of KLF4 in terminally differentiated intestinal cells led to an increase in goblet cells, implying a role for KLF4 in maintaining goblet cell population and mispositioning of Paneth cells, suggesting KLF4-dependent localization [
61].
3. Krüppel-like Factor 5
3.1. Homeostasis
KLF5 is also known as Intestinal Krüppel-like factor (IKLF) due to its high expression in intestinal epithelium. However, KLF5 can be detected in almost all tissues, including breast, prostate, pancreas, intestine, lung, bladder, and skeletal muscle [
22,
32,
60,
149,
150,
151]. KLF5 regulates many cellular processes, including cell cycle, proliferation, migration, invasion, stemness, apoptosis, and autophagy, and plays a crucial role in maintaining gut homeostasis [
32,
152,
153,
154,
155,
156,
157,
158,
159,
160,
161,
162,
163]. Notably, KLF5 regulates villus formation and initiates cytodifferentiation in embryonic intestinal epithelium. Deletion of
Klf5 from intestinal epithelium during embryogenesis leads to downregulation of multiple genes such as E74-like ETS transcription factor 3 (
Elf3),
Pparg, Atonal BHLH transcription factor 1 (
Atoh1), Achaete-scute family bHLH transcription factor 2 (
Ascl2), Hepatocyte nuclear factor 4 alpha (
Hnf4a), Neurogenin 3 (
Neurog3), and Caudal Type Homeobox 1 (
Cdx1) [
164].
Similarly, data obtained from
Klf5 deletion in the gut suggest that KLF5 plays a role in maintaining epithelial proliferation, differentiation, and cell positioning along the crypt radial axis in adult mice [
165,
166]. Mice with deletion of
Klf5 within active intestinal epithelial stem cells have decreased expression of intestinal stem cell signature genes, such as
Lgr5, Olfactomedin 4 (
Olfm4), and
Ascl2, and impaired stem cell renewal. KLF5 is crucial for stem cell activity and regeneration of the intestinal epithelium after injury [
167,
168]. KLF5 also regulates DNA damage repair in intestinal epithelial cells upon radiation injury. In mice with heterozygous deletion of
Klf5 in intestinal epithelial cells, genes involved in nucleotide excision repair, mismatch repair, and non-homologous end-joining were significantly downregulated compared to wild-type mice [
169]. Mice with intestinal epithelium-specific deletion of
Klf5 also developed a Th-17-mediated immune response and subsequent colitis, suggesting a protective role of KLF5 against intestinal inflammation [
170].
Evidently, KLF5 is indicated in a wide range of processes to ensure intestinal epithelial homeostasis in the presence of insults. While the lack of KLF5 activity can lead to insufficient self-renewal and intestinal integrity, overactivation of KLF5 may cause uncontrolled cell proliferation and differentiation, ultimately leading to tumorigenesis. As such, understanding the role of KLF5 in achieving balance in these cellular processes is essential to ensure intestinal health. However, whether KLF5 functions to upregulate or downregulate these processes is context-dependent and highly controversial.
3.2. Colorectal Cancer
3.2.1. KLF5 Is a Pro-Proliferative Factor in CRC
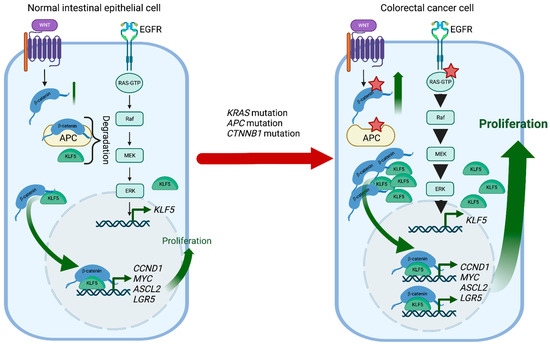
KLF5 is a pro-proliferative transcription factor downstream of the classical Mitogen-activated protein kinase (MAPK-ERK-RAS) pathway and directly regulated by Early Growth Response 1 (EGR1) [
171]. Activation of the KRAS oncogene plays an essential role in CRC pathophysiology, and KLF5 contributes to colorectal tumorigenesis induced by a constitutively activating KRAS mutation (G12V) (
Figure 2). For example, Klf5 haploinsufficiency in Apc
Min/+/Kras
G12V mice resulted in significantly reduced tumor number and size compared to Apc
Min/+ mice [
172]. In addition, increased levels of KLF5 were observed in spontaneous hyperplastic intestinal polyp development and colonic tumorigenesis in Villin-Cre/LSL-KRAS
G12D mice, further supporting KLF5′s role as a mediator of the KRAS pathway in CRC formation [
173]. Interestingly, while the Villin-Cre/LSL-KRAS
G12D mice displayed decreased survival when treated with AOM compared to controls, loss of one Klf5 allele showed reduced levels of KRAS effector proteins and, as a result, reduced mortality upon AOM treatment [
173]. Overall, KLF5 expression appears essential in exerting the oncogenic, pro-proliferative effects of KRAS mutations in CRC.
Figure 2. KLF5 and WNT signaling in CRC development. In normal intestinal epithelial cells, KLF5 and β-catenin are regulated by multiple mechanisms such as degradation or well-coordinated WNT and MAPK kinase activation. During CRC development,
Kras mutations increase
KLF5 expression, while
Apc and
Ctnnb1 mutations increase WNT pathway activity by increasing the stability and transcriptional activity of β-catenin. In the context of these mutations, KLF5 and β-catenin contribute to CRC tumorigenesis by inducing transcription of multiple genes such as
Ccnd1,
c-Myc,
Ascl2, or
Lgr5. Red stars mark mutations. Created with
BioRender.com (accessed on 12 April 2023).
The HIPPO pathway regulates cell stemness and proliferation via two key transcriptional coactivators, Yes1 Associated Transcriptional Regulator (YAP1) and WW domain-containing transcription regulator protein 1 (TAZ). The KLF5-YAP1 complex induces transcription of
Ascl2, a WNT signaling target, to ensure the self-renewability of CRC progenitor cells [
158]. Synaptopodin-2 (SYNPO2) was shown to inhibit the KLF5-YAP signaling pathway and suppress hypoxia-induced progression of CRC [
174].
The TGF-β/SMAD4-signaling pathway and its role in CRC are well-established. Silencing
KLF5 was found to sensitize SMAD4-deficient cells to TGF-β-induced apoptosis. Conversely, overexpression of
KLF5 significantly inhibited TGF-β-induced apoptosis in SMAD4-proficient cells. This suggests that KLF5 acts as an oncogene in CRC regardless of SMAD4 expression [
175]. One recent study discovered that primary mesenchymal stromal cells (MSCs) play a dual role in regulating C-X-C Motif Chemokine Ligand 5 (CXCL5), which is significantly overexpressed in CRC, allowing for distant metastasis and angiogenesis. MSCs not only secreted C-C Motif Chemokine Ligand 7 (CCL7) to promote acetylation of KLF5 and upregulate transcription of CXCL5 but also secreted TGF-β to regulate SMAD4 and reverse the effect of KLF5 on the transcription of CXCL5 [
176].
3.2.2. KLF5-WNT/β-Catenin Positive Feedback Loop Regulated CRC Development and Progression
Germline loss-of-function mutation of
APC and mutations in
CTNNB1 have been identified as a cause of colorectal cancer. APC plays an essential role in regulating the activity of β-catenin, which controls the WNT signaling pathway responsible for maintaining the proliferation of the intestinal crypt epithelium. KLF5 is a crucial mediator of these interactions contributing to CRC tumorigenesis.
Klf5 haploinsufficiency in the context of
Apc mutation was associated with lower levels and reduced nuclear localization of β-catenin, resulting in reduced expression of
Ccnd1 and
c-Myc, downregulation of the WNT pathway activity, and decreased polyp formation [
177]. In addition, the formation of lethal colorectal adenomas and carcinomas induced by β-catenin mutations in Lgr5
+ stem cells was entirely suppressed by
Klf5 deletion [
178]. Overall, lack of KLF5 expression prevented the tumorigenic effects of
Apc mutation and β-catenin activation, suggesting the oncogenic function and necessity of KLF5 in CRC (
Figure 2).
Lysophosphatidic acid (LPA), a simple phospholipid with potent mitogenic effects, and its receptor LPAR modulate the tumorigenic effects of
APC mutation. Compared to
ApcMin/+ mice,
ApcMin/+/Lpar2−/− mice exhibited decreased tumor progression and hypoxia in response to reduced expression of
Klf5,
Ctnnb1,
Ccnd1 and
c-Myc [
179]. A recent study proposed a new mechanism by which KLF5 modulates the WNT/β-catenin pathway in the presence of LPA. Contrary to previous findings, silencing KLF5 did not alter the nuclear translocation of β-catenin by LPA. Instead, KLF5 was found to facilitate LPA-induced formation and transcriptional activity of the β-catenin/TCF complex to promote colon cancer cell proliferation [
180].
Ketogenesis is significantly decreased in the tumor microenvironment of CRC. As such, a ketogenic diet of high lipids and low carbohydrates has been recommended for cancer patients. Increasing ketogenesis markedly decreased KLF5-dependent synthesis of C-X-C Motif Chemokine Ligand 12 (CXCL12) in cancer-associated fibroblasts, ultimately increasing the infiltration of immune effector cells in tumors and enhancing sensitivity to immune checkpoint inhibitors specific for programmed cell death 1 (PD-1) [
181]. By the same mechanism, increasing ketogenesis inhibited CRC migration, invasion, and metastasis both in vitro and in vivo [
182].
3.2.3. KLF5 and microRNA in CRC
MiRs bind directly to the 3′UTR of KLF5, thereby suppressing colorectal cancer cell proliferation, migration, and stemness in vitro and inhibiting tumor growth in vivo in mouse models. Recent studies have found several miRs that target and modulate KLF5 at the post-transcriptional level to regulate the development of CRC.
MiR-143 and miR-145 have been found to decrease the expression of
KLF5 in CRC [
183]. Consistent with this finding, one study suggests that increased expression of the long intergenic noncoding RNA (lncRNA) LINC00908 may act as a competing endogenous RNA to negatively regulate the miR-143-3p/KLF5 axis, thereby promoting cell proliferation and survival of colorectal cancer cells [
185]. miR-4711-5p was also shown to bind directly to the 3′UTR of
KLF5, thereby suppressing colorectal cancer cell proliferation, migration, and stemness in vitro and inhibiting tumor growth in vivo in mouse models [
186]. Overexpression of miR-143-3p was also associated with downregulation of
KLF5 and was detected in significantly lower amounts in more advanced CRC [
187].
In addition, lncRNAs have been identified as targets of KLF5 in CRC. For example, lncRNA plasmacytoma variant translocation 1 (PVT1) was found to be regulated by its upstream transcription factor KLF5 and was detected in significant amounts in CRC [
188]. Small Nucleolar RNA Host Gene 12 (SNHG12) was also proposed as a lncRNA target for KLF5, positively regulating CRC invasion and distal metastasis. However, whether targeting KLF5-SNHG12 will produce therapeutic benefits is still being investigated [
189]. Another study proposes a novel mechanism by which the KLF5 protein constructs a loop-like three-dimensional genome structure consisting of
KLF5 promoter, enhancer, and the transcription start site region of Colon Cancer Associated Transcript 1 (CCAT1). This promoter-enhancer loop may modulate the expression of KLF5 and CCAT1, resulting in the maintenance of colorectal cancer stemness [
157]. Recently, low-molecular-weight compounds targeting the hydrophobic α-helix structure of KLF5, known as a potential interface for protein–protein interaction, were synthesized using pyrazinooxadiazine-4,7-dione. Once bound to this interface, these compounds selectively suppressed levels of the KLF5 protein and reduced the expression of proteins involved in the WNT signaling pathway, thereby inhibiting the proliferation and survival of transplanted colorectal cancer cells in vivo [
190].
3.2.4. KLF5 as a Therapeutic Target in CRC
Using an ultra-high-throughput screen, our group identified two KLF5-selective compounds, CID 439501 and 5951923, that significantly decrease endogenous KLF5 protein levels and reduce the viability of several CRC cell lines [
191]. A small-molecule compound called ML264 was found to be a KLF5 inhibitor, preventing the expression of
KLF5 and the growth of CRC xenograft tumors [
192]. ML264 exerted this effect by inhibiting the RAS/MAPK/PI3K and the WNT/β-catenin signaling pathway. The same KLF5 inhibitor was recently used to investigate CRC resistance to oxaliplatin, a first-line chemotherapy drug commonly used in CRC. Using ML264, the study successfully inhibited the KLF5/BCL-2/Caspase 3 signaling pathway, thereby restoring the apoptotic response and significantly restoring sensitivity to oxaliplatin in CRC patient-derived organoids [
193]. Interestingly, SR18662, a derivative of ML264, demonstrated enhanced abilities to inhibit KLF5, the MAPK and WNT pathways, and the growth of CRC in vitro and in vivo with the ability to exert cytotoxic effects [
194]. Dual-specificity phosphatase 10 (DUSP10), known for its role in deactivating MAP kinases, reduced intestinal epithelial cell proliferation via inhibition of ERK1/2 activation and KLF5 expression [
195].
KLF5 also modulates CRC response to radiation therapy. For example, HCT116 cells with significantly higher levels of KLF5 were shown to increase CyclinD1 and β-catenin and promote better cell viability than control cells when subjected to radiation therapy [
199]. In addition, the depletion of KLF5 in HCT116 cells increased CRC sensitivity to DNA-damaging ultraviolet irradiation therapy by failing to induce the proto-oncogene, serine/threonine kinase 1 (PIM1) survival kinase [
200]. It appears that overexpression of
KLF5 confers resistance to radiotherapy, while reduction of KLF5 may increase susceptibility to radiotherapeutic effects in CRC.
3.2.5. KLF5 as a Biomarker of CRC
Overexpression of
KLF5 may be used as a predictive biomarker for poor tumor regression after preoperative chemoradiation therapy, the standard treatment for locally advanced rectal cancer [
199]. A recent study was the first to examine the expression of levels of KLF5 in patients with colorectal cancer to determine correlation with clinical outcomes. The study revealed that high expression of KLF5 in tissues collected from CRC patients was associated with vascular invasion, increased serum carbohydrate 19-9, larger metastatic liver tumors, and poorer prognosis after surgery. While further investigation is needed, KLF5 upregulation of
c-MYC and
CCND1 via promoter binding may be the mechanism underlining these effects. Thus, high KLF5 expression can independently predict poor prognosis in patients with primary CRC and liver metastasis [
201]. However, KLF5 and its use as a prognosis marker in CRC must be studied further.