ETS transcription factors are a highly conserved family of proteins involved in the progression of many cancers, such as breast and prostate carcinomas, Ewing’s sarcoma, and leukaemias. This significant involvement can be explained by their roles at all stages of carcinogenesis progression. Generally, their expression in tumours is associated with a poor prognosis and an aggressive phenotype. Until now, no efficient therapeutic strategy had emerged to specifically target ETS-expressing tumours. Nevertheless, there is evidence that pharmacological inhibition of poly(ADP-ribose) polymerase-1 (PARP-1), a key DNA repair enzyme, specifically sensitises ETS-expressing cancer cells to DNA damage and limits tumour progression by leading some of the cancer cells to death. These effects result from a strong interplay between ETS transcription factors and the PARP-1 enzyme.
- ETS transcription factors
- PARP-1
- pharmacological inhibition
- cancer therapy
- DNA damage
1. Introduction
2. ETS Transcription Factors Expression in Cancers
2.1. Expression and Involvement in Cancers
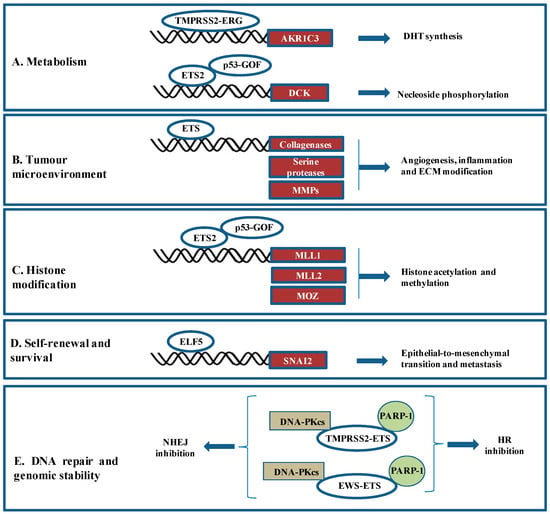
2.2. ETS Fusions and Cancers
3. PARP-1 Inhibition in Cancer Therapy
3.1. The Plethoric Roles of PARP-1 in Cancer Cells
3.2. PARP-1 Inhibition in Cancer Cells and Clinical Trials
3.3. Limitation of PARP-1 Inhibitors in Cancer Therapy
4. Molecular Interplay between ETS Transcription Factors and PARP-1 Enzyme
4.1. Regulation of PARP-1 Expression and Activity by ETS Transcription Factors
4.2. Control of ETS Transcription Factors Functions by PARP-1
5. Cellular Consequences of PARP-1 Inhibition on ETS-Expressing Tumour Cells
5.1. PARP-1 Inhibition Slows down ETS-Expressing Tumour Growth by Inhibiting Invasion and Metastasis and Decreasing Cell Survival
5.2. PARP-1 Inhibition Causes the Accumulation of Unrepaired DSB in ETS-Expressing Cells
This entry is adapted from the peer-reviewed paper 10.3390/ijms241713454
References
- Degnan, B.M.; Degnan, S.M.; Naganuma, T.; Morse, D.E. The ets Multigene Family Is Conserved throughout the Metazoa. Nucl. Acids Res. 1993, 21, 3479–3484.
- Laudet, V.; Hänni, C.; Stéhelin, D.; Duterque-Coquillaud, M. Molecular Phylogeny of the ETS Gene Family. Oncogene 1999, 18, 1351–1359.
- Hollenhorst, P.C.; McIntosh, L.P.; Graves, B.J. Genomic and Biochemical Insights into the Specificity of ETS Transcription Factors. Annu. Rev. Biochem. 2011, 80, 437–471.
- Wei, G.-H.; Badis, G.; Berger, M.F.; Kivioja, T.; Palin, K.; Enge, M.; Bonke, M.; Jolma, A.; Varjosalo, M.; Gehrke, A.R.; et al. Genome-Wide Analysis of ETS-Family DNA-Binding in Vitro and in Vivo. EMBO J. 2010, 29, 2147–2160.
- Oikawa, T.; Yamada, T. Molecular Biology of the Ets Family of Transcription Factors. Gene 2003, 303, 11–34.
- Seth, A.; Watson, D.K. ETS Transcription Factors and Their Emerging Roles in Human Cancer. Eur. J. Cancer 2005, 41, 2462–2478.
- Hsu, T.; Trojanowska, M.; Watson, D.K. Ets Proteins in Biological Control and Cancer. J. Cell Biochem. 2004, 91, 896–903.
- Shao, L.; Tekedereli, I.; Wang, J.; Yuca, E.; Tsang, S.; Sood, A.; Lopez-Berestein, G.; Ozpolat, B.; Ittmann, M. Highly Specific Targeting of the TMPRSS2/ERG Fusion Gene Using Liposomal Nanovectors. Clin. Cancer Res. 2012, 18, 6648–6657.
- Li, L.; Hobson, L.; Perry, L.; Clark, B.; Heavey, S.; Haider, A.; Sridhar, A.; Shaw, G.; Kelly, J.; Freeman, A.; et al. Targeting the ERG Oncogene with Splice-Switching Oligonucleotides as a Novel Therapeutic Strategy in Prostate Cancer. Br. J. Cancer 2020, 123, 1024–1032.
- Laitem, C.; Leprivier, G.; Choul-Li, S.; Begue, A.; Monte, D.; Larsimont, D.; Dumont, P.; Duterque-Coquillaud, M.; Aumercier, M. Ets-1 P27: A Novel Ets-1 Isoform with Dominant-Negative Effects on the Transcriptional Properties and the Subcellular Localization of Ets-1 P51. Oncogene 2009, 28, 2087–2099.
- Sahin, A.; Vercamer, C.; Kaminski, A.; Fuchs, T.; Florin, A.; Hahne, J.C.; Mattot, V.; Pourtier-Manzanedo, A.; Pietsch, T.; Fafeur, V.; et al. Dominant-Negative Inhibition of Ets 1 Suppresses Tumor Growth, Invasion and Migration in Rat C6 Glioma Cells and Reveals Differentially Expressed Ets 1 Target Genes. Int. J. Oncol. 2009, 34, 377–389.
- Wang, X.; Qiao, Y.; Asangani, I.A.; Ateeq, B.; Poliakov, A.; Cieślik, M.; Pitchiaya, S.; Chakravarthi, B.V.S.K.; Cao, X.; Jing, X.; et al. Development of Peptidomimetic Inhibitors of the ERG Gene Fusion Product in Prostate Cancer. Cancer Cell 2017, 31, 532–548.e7.
- Nhili, R.; Peixoto, P.; Depauw, S.; Flajollet, S.; Dezitter, X.; Munde, M.M.; Ismail, M.A.; Kumar, A.; Farahat, A.A.; Stephens, C.E.; et al. Targeting the DNA-Binding Activity of the Human ERG Transcription Factor Using New Heterocyclic Dithiophene Diamidines. Nucleic Acids Res. 2013, 41, 125–138.
- Harlow, M.L.; Chasse, M.H.; Boguslawski, E.A.; Sorensen, K.M.; Gedminas, J.M.; Kitchen-Goosen, S.M.; Rothbart, S.B.; Taslim, C.; Lessnick, S.L.; Peck, A.S.; et al. Trabectedin Inhibits EWS-FLI1 and Evicts SWI/SNF from Chromatin in a Schedule-Dependent Manner. Clin. Cancer Res. 2019, 25, 3417–3429.
- Harlow, M.L.; Maloney, N.; Roland, J.; Guillen Navarro, M.J.; Easton, M.K.; Kitchen-Goosen, S.M.; Boguslawski, E.A.; Madaj, Z.B.; Johnson, B.K.; Bowman, M.J.; et al. Lurbinectedin Inactivates the Ewing Sarcoma Oncoprotein EWS-FLI1 by Redistributing It within the Nucleus. Cancer Res. 2016, 76, 6657–6668.
- Erkizan, H.V.; Kong, Y.; Merchant, M.; Schlottmann, S.; Barber-Rotenberg, J.S.; Yuan, L.; Abaan, O.D.; Chou, T.-H.; Dakshanamurthy, S.; Brown, M.L.; et al. A Small Molecule Blocking Oncogenic Protein EWS-FLI1 Interaction with RNA Helicase A Inhibits Growth of Ewing’s Sarcoma. Nat. Med. 2009, 15, 750–756.
- Rosati, R.; Polin, L.; Ducker, C.; Li, J.; Bao, X.; Selvakumar, D.; Kim, S.; Xhabija, B.; Larsen, M.; McFall, T.; et al. Strategy for Tumor-Selective Disruption of Androgen Receptor Function in the Spectrum of Prostate Cancer. Clin. Cancer Res. 2018, 24, 6509–6522.
- Butler, M.S.; Roshan-Moniri, M.; Hsing, M.; Lau, D.; Kim, A.; Yen, P.; Mroczek, M.; Nouri, M.; Lien, S.; Axerio-Cilies, P.; et al. Discovery and Characterization of Small Molecules Targeting the DNA-Binding ETS Domain of ERG in Prostate Cancer. Oncotarget 2017, 8, 42438–42454.
- Rahim, S.; Beauchamp, E.M.; Kong, Y.; Brown, M.L.; Toretsky, J.A.; Üren, A. YK-4-279 Inhibits ERG and ETV1 Mediated Prostate Cancer Cell Invasion. PLoS ONE 2011, 6, e19343.
- Rahim, S.; Minas, T.; Hong, S.-H.; Justvig, S.; Çelik, H.; Kont, Y.S.; Han, J.; Kallarakal, A.T.; Kong, Y.; Rudek, M.A.; et al. A Small Molecule Inhibitor of ETV1, YK-4-279, Prevents Prostate Cancer Growth and Metastasis in a Mouse Xenograft Model. PLoS ONE 2014, 9, e114260.
- Pop, M.S.; Stransky, N.; Garvie, C.W.; Theurillat, J.-P.; Hartman, E.C.; Lewis, T.A.; Zhong, C.; Culyba, E.K.; Lin, F.; Daniels, D.S.; et al. A Small Molecule That Binds and Inhibits the ETV1 Transcription Factor Oncoprotein. Mol. Cancer Ther. 2014, 13, 1492–1502.
- Liu, T.; Xia, L.; Yao, Y.; Yan, C.; Fan, Y.; Gajendran, B.; Yang, J.; Li, Y.-J.; Chen, J.; Filmus, J.; et al. Identification of Diterpenoid Compounds That Interfere with Fli-1 DNA Binding to Suppress Leukemogenesis. Cell Death Dis. 2019, 10, 117.
- Liu, Y.; Eckenrode, J.M.; Zhang, Y.; Zhang, J.; Hayden, R.C.; Kyomuhangi, A.; Ponomareva, L.V.; Cui, Z.; Rohr, J.; Tsodikov, O.V.; et al. Mithramycin 2′-Oximes with Improved Selectivity, Pharmacokinetics, and Ewing Sarcoma Antitumor Efficacy. J. Med. Chem. 2020, 63, 14067–14086.
- Mitra, P.; Eckenrode, J.M.; Mandal, A.; Jha, A.K.; Salem, S.M.; Leggas, M.; Rohr, J. Development of Mithramycin Analogues with Increased Selectivity toward ETS Transcription Factor Expressing Cancers. J. Med. Chem. 2018, 61, 8001–8016.
- Mohamed, A.A.; Xavier, C.P.; Sukumar, G.; Tan, S.-H.; Ravindranath, L.; Seraj, N.; Kumar, V.; Sreenath, T.; McLeod, D.G.; Petrovics, G.; et al. Identification of a Small Molecule That Selectively Inhibits ERG-Positive Cancer Cell Growth. Cancer Res. 2018, 78, 3659–3671.
- Grohar, P.J.; Glod, J.; Peer, C.J.; Sissung, T.M.; Arnaldez, F.I.; Long, L.; Figg, W.D.; Whitcomb, P.; Helman, L.J.; Widemann, B.C. A Phase I/II Trial and Pharmacokinetic Study of Mithramycin in Children and Adults with Refractory Ewing Sarcoma and EWS-FLI1 Fusion Transcript. Cancer Chemother. Pharmacol. 2017, 80, 645–652.
- Brenner, J.C.; Feng, F.Y.; Han, S.; Patel, S.; Goyal, S.V.; Bou-Maroun, L.M.; Liu, M.; Lonigro, R.; Prensner, J.R.; Tomlins, S.A.; et al. PARP-1 Inhibition as a Targeted Strategy to Treat Ewing’s Sarcoma. Cancer Res. 2012, 72, 1608–1613.
- Brenner, J.C.; Ateeq, B.; Li, Y.; Yocum, A.K.; Cao, Q.; Asangani, I.A.; Patel, S.; Wang, X.; Liang, H.; Yu, J.; et al. Mechanistic Rationale for Inhibition of Poly(ADP-Ribose) Polymerase in ETS Gene Fusion-Positive Prostate Cancer. Cancer Cell 2011, 19, 664–678.
- Legrand, A.J.; Choul-Li, S.; Spriet, C.; Idziorek, T.; Vicogne, D.; Drobecq, H.; Dantzer, F.; Villeret, V.; Aumercier, M. The Level of Ets-1 Protein Is Regulated by Poly(ADP-Ribose) Polymerase-1 (PARP-1) in Cancer Cells to Prevent DNA Damage. PLoS ONE 2013, 8, e55883.
- Gelmon, K.A.; Tischkowitz, M.; Mackay, H.; Swenerton, K.; Robidoux, A.; Tonkin, K.; Hirte, H.; Huntsman, D.; Clemons, M.; Gilks, B.; et al. Olaparib in Patients with Recurrent High-Grade Serous or Poorly Differentiated Ovarian Carcinoma or Triple-Negative Breast Cancer: A Phase 2, Multicentre, Open-Label, Non-Randomised Study. Lancet Oncol. 2011, 12, 852–861.
- Loap, P.; Loirat, D.; Berger, F.; Cao, K.; Ricci, F.; Jochem, A.; Raizonville, L.; Mosseri, V.; Fourquet, A.; Kirova, Y. Combination of Olaparib with Radiotherapy for Triple-negative Breast Cancers: One-year Toxicity Report of the RADIOPARP Phase I Trial. Int. J. Cancer 2021, 149, 1828–1832.
- Wu, Y.; Xu, S.; Cheng, S.; Yang, J.; Wang, Y. Clinical Application of PARP Inhibitors in Ovarian Cancer: From Molecular Mechanisms to the Current Status. J. Ovarian Res. 2023, 16, 6.
- Leprince, D.; Gegonne, A.; Coll, J.; de Taisne, C.; Schneeberger, A.; Lagrou, C.; Stehelin, D. A Putative Second Cell-Derived Oncogene of the Avian Leukaemia Retrovirus E26. Nature 1983, 306, 395–397.
- Nunn, M.F.; Seeburg, P.H.; Moscovici, C.; Duesberg, P.H. Tripartite Structure of the Avian Erythroblastosis Virus E26 Transforming Gene. Nature 1983, 306, 391–395.
- Radke, K.; Beug, H.; Kornfeld, S.; Graf, T. Transformation of Both Erythroid and Myeloid Cells by E26, an Avian Leukemia Virus That Contains the Myb Gene. Cell 1982, 31, 643–653.
- Hsing, M.; Wang, Y.; Rennie, P.S.; Cox, M.E.; Cherkasov, A. ETS Transcription Factors as Emerging Drug Targets in Cancer. Med. Res. Rev. 2020, 40, 413–430.
- Powell, K.; Semaan, L.; Conley-LaComb, M.K.; Asangani, I.; Wu, Y.-M.; Ginsburg, K.B.; Williams, J.; Squire, J.A.; Maddipati, K.R.; Cher, M.L.; et al. ERG/AKR1C3/AR Constitutes a Feed-Forward Loop for AR Signaling in Prostate Cancer Cells. Clin. Cancer Res. 2015, 21, 2569–2579.
- Kollareddy, M.; Dimitrova, E.; Vallabhaneni, K.C.; Chan, A.; Le, T.; Chauhan, K.M.; Carrero, Z.I.; Ramakrishnan, G.; Watabe, K.; Haupt, Y.; et al. Regulation of Nucleotide Metabolism by Mutant P53 Contributes to Its Gain-of-Function Activities. Nat. Commun. 2015, 6, 7389.
- Kar, A.; Gutierrez-Hartmann, A. Molecular Mechanisms of ETS Transcription Factor-Mediated Tumorigenesis. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 522–543.
- Zhu, J.; Sammons, M.A.; Donahue, G.; Dou, Z.; Vedadi, M.; Getlik, M.; Barsyte-Lovejoy, D.; Al-awar, R.; Katona, B.W.; Shilatifard, A.; et al. Gain-of-Function P53 Mutants Co-Opt Chromatin Pathways to Drive Cancer Growth. Nature 2015, 525, 206–211.
- Chakrabarti, R.; Hwang, J.; Andres Blanco, M.; Wei, Y.; Lukačišin, M.; Romano, R.-A.; Smalley, K.; Liu, S.; Yang, Q.; Ibrahim, T.; et al. Elf5 Inhibits the Epithelial–Mesenchymal Transition in Mammary Gland Development and Breast Cancer Metastasis by Transcriptionally Repressing Snail2. Nat. Cell Biol. 2012, 14, 1212–1222.
- Chatterjee, P.; Choudhary, G.S.; Alswillah, T.; Xiong, X.; Heston, W.D.; Magi-Galluzzi, C.; Zhang, J.; Klein, E.A.; Almasan, A. The TMPRSS2–ERG Gene Fusion Blocks XRCC4-Mediated Nonhomologous End-Joining Repair and Radiosensitizes Prostate Cancer Cells to PARP Inhibition. Mol. Cancer Ther. 2015, 14, 1896–1906.
- Lessnick, S.L.; Ladanyi, M. Molecular Pathogenesis of Ewing Sarcoma: New Therapeutic and Transcriptional Targets. Annu. Rev. Pathol. Mech. Dis. 2012, 7, 145–159.
- Tomlins, S.A.; Bjartell, A.; Chinnaiyan, A.M.; Jenster, G.; Nam, R.K.; Rubin, M.A.; Schalken, J.A. ETS Gene Fusions in Prostate Cancer: From Discovery to Daily Clinical Practice. Eur. Urol. 2009, 56, 275–286.
- Qian, C.; Li, D.; Chen, Y. ETS Factors in Prostate Cancer. Cancer Lett. 2022, 530, 181–189.
- De Braekeleer, E.; Douet-Guilbert, N.; Morel, F.; Le Bris, M.-J.; Basinko, A.; De Braekeleer, M. ETV6 Fusion Genes in Hematological Malignancies: A Review. Leuk. Res. 2012, 36, 945–961.
- Apfelbaum, A.A.; Wrenn, E.D.; Lawlor, E.R. The Importance of Fusion Protein Activity in Ewing Sarcoma and the Cell Intrinsic and Extrinsic Factors That Regulate It: A Review. Front. Oncol. 2022, 12, 1044707.
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.-W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent Fusion of TMPRSS2 and ETS Transcription Factor Genes in Prostate Cancer. Science 2005, 310, 644–648.
- Khosh Kish, E.; Choudhry, M.; Gamallat, Y.; Buharideen, S.M.; Bismar, T.A. The Expression of Proto-Oncogene ETS-Related Gene (ERG) Plays a Central Role in the Oncogenic Mechanism Involved in the Development and Progression of Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 4772.
- Nicholas, T.R.; Strittmatter, B.G.; Hollenhorst, P.C. Oncogenic ETS Factors in Prostate Cancer. In Prostate Cancer; Dehm, S.M., Tindall, D.J., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2019; Volume 1210, pp. 409–436. ISBN 978-3-030-32655-5.
- Kim, M.Y.; Zhang, T.; Kraus, W.L. Poly(ADP-Ribosyl)Ation by PARP-1: ‘PAR-Laying’ NAD + into a Nuclear Signal. Genes Dev. 2005, 19, 1951–1967.
- De Vos, M.; Schreiber, V.; Dantzer, F. The Diverse Roles and Clinical Relevance of PARPs in DNA Damage Repair: Current State of the Art. Biochem. Pharmacol. 2012, 84, 137–146.
- Ray Chaudhuri, A.; Nussenzweig, A. The Multifaceted Roles of PARP1 in DNA Repair and Chromatin Remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621.
- Luo, X.; Kraus, W.L. On PAR with PARP: Cellular Stress Signaling through Poly(ADP-Ribose) and PARP-1. Genes Dev. 2012, 26, 417–432.
- Kraus, W.L. Transcriptional Control by PARP-1: Chromatin Modulation, Enhancer-Binding, Coregulation, and Insulation. Curr. Opin. Cell Biol. 2008, 20, 294–302.
- Ogino, H.; Nozaki, T.; Gunji, A.; Maeda, M.; Suzuki, H.; Ohta, T.; Murakami, Y.; Nakagama, H.; Sugimura, T.; Masutani, M. Loss of Parp-1 Affects Gene Expression Profile in a Genome-Wide Manner in ES Cells and Liver Cells. BMC Genom. 2007, 8, 41.
- Masutani, M.; Nakagama, H.; Sugimura, T. Poly(ADP-Ribosyl)Ation in Relation to Cancer and Autoimmune Disease. Cell Mol. Life Sci. 2005, 62, 769–783.
- Akanksha; Mishra, S.; Kar, A.; Karthik, J.; Srivastava, A.; Khanna, R.; Meena, R. Expression of Poly(Adenosine Diphosphate-Ribose) Polymerase Protein in Breast Cancer. J. Mid-Life Health 2022, 13, 213.
- Bièche, I.; de Murcia, G.; Lidereau, R. Poly(ADP-Ribose) Polymerase Gene Expression Status and Genomic Instability in Human Breast Cancer. Clin. Cancer Res. 1996, 2, 1163–1167.
- Quiles-Perez, R.; Muñoz-Gámez, J.A.; Ruiz-Extremera, Á.; O’Valle, F.; Sanjuán-Nuñez, L.; Martín-Álvarez, A.B.; Martín-Oliva, D.; Caballero, T.; Muñoz de Rueda, P.; León, J.; et al. Inhibition of Poly Adenosine Diphosphate-Ribose Polymerase Decreases Hepatocellular Carcinoma Growth by Modulation of Tumor-Related Gene Expression. Hepatology 2010, 51, 255–266.
- Nosho, K.; Yamamoto, H.; Mikami, M.; Taniguchi, H.; Takahashi, T.; Adachi, Y.; Imamura, A.; Imai, K.; Shinomura, Y. Overexpression of Poly(ADP-Ribose) Polymerase-1 (PARP-1) in the Early Stage of Colorectal Carcinogenesis. Eur. J. Cancer 2006, 42, 2374–2381.
- Staibano, S.; Pepe, S.; Muzio, L.L.; Somma, P.; Mascolo, M.; Argenziano, G.; Scalvenzi, M.; Salvatore, G.; Fabbrocini, G.; Molea, G.; et al. Poly(Adenosine Diphosphate-Ribose) Polymerase 1 Expression in Malignant Melanomas from Photoexposed Areas of the Head and Neck Region. Hum. Pathol. 2005, 36, 724–731.
- Masutani, M.; Fujimori, H. Poly(ADP-Ribosyl)Ation in Carcinogenesis. Mol. Asp. Med. 2013, 34, 1202–1216.
- Zaremba, T.; Ketzer, P.; Cole, M.; Coulthard, S.; Plummer, E.R.; Curtin, N.J. Poly(ADP-Ribose) Polymerase-1 Polymorphisms, Expression and Activity in Selected Human Tumour Cell Lines. Br. J. Cancer 2009, 101, 256–262.
- Dias, M.P.; Moser, S.C.; Ganesan, S.; Jonkers, J. Understanding and Overcoming Resistance to PARP Inhibitors in Cancer Therapy. Nat. Rev. Clin. Oncol. 2021, 18, 773–791.
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834.
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.H.; Zander, S.A.L.; Derksen, P.W.B.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High Sensitivity of BRCA1-Deficient Mammary Tumors to the PARP Inhibitor AZD2281 Alone and in Combination with Platinum Drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084.
- Jaspers, J.E.; Kersbergen, A.; Boon, U.; Sol, W.; Van Deemter, L.; Zander, S.A.; Drost, R.; Wientjens, E.; Ji, J.; Aly, A.; et al. Loss of 53BP1 Causes PARP Inhibitor Resistance in Brca1-Mutated Mouse Mammary Tumors. Cancer Discov. 2013, 3, 68–81.
- Gogola, E.; Duarte, A.A.; de Ruiter, J.R.; Wiegant, W.W.; Schmid, J.A.; de Bruijn, R.; James, D.I.; Guerrero Llobet, S.; Vis, D.J.; Annunziato, S.; et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell 2018, 33, 1078–1093.e12.
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-Wide and High-Density CRISPR-Cas9 Screens Identify Point Mutations in PARP1 Causing PARP Inhibitor Resistance. Nat. Commun. 2018, 9, 1849.
- Johnson, N.; Johnson, S.F.; Yao, W.; Li, Y.-C.; Choi, Y.-E.; Bernhardy, A.J.; Wang, Y.; Capelletti, M.; Sarosiek, K.A.; Moreau, L.A.; et al. Stabilization of Mutant BRCA1 Protein Confers PARP Inhibitor and Platinum Resistance. Proc. Natl. Acad. Sci. USA 2013, 110, 17041–17046.
- Ter Brugge, P.; Kristel, P.; Van Der Burg, E.; Boon, U.; De Maaker, M.; Lips, E.; Mulder, L.; De Ruiter, J.; Moutinho, C.; Gevensleben, H.; et al. Mechanisms of Therapy Resistance in Patient-Derived Xenograft Models of BRCA1-Deficient Breast Cancer. JNCI J. Natl. Cancer Inst. 2016, 108, djw148.
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.-Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of All BRCA1 Copies Predicts Response to the PARP Inhibitor Rucaparib in Ovarian Carcinoma. Nat. Commun. 2018, 9, 3970.
- Nacson, J.; Krais, J.J.; Bernhardy, A.J.; Clausen, E.; Feng, W.; Wang, Y.; Nicolas, E.; Cai, K.Q.; Tricarico, R.; Hua, X.; et al. BRCA1 Mutation-Specific Responses to 53BP1 Loss-Induced Homologous Recombination and PARP Inhibitor Resistance. Cell Rep. 2018, 25, 1384.
- Dev, H.; Chiang, T.-W.W.; Lescale, C.; de Krijger, I.; Martin, A.G.; Pilger, D.; Coates, J.; Sczaniecka-Clift, M.; Wei, W.; Ostermaier, M.; et al. Shieldin Complex Promotes DNA End-Joining and Counters Homologous Recombination in BRCA1-Null Cells. Nat. Cell Biol. 2018, 20, 954–965.
- Tomida, J.; Takata, K.; Bhetawal, S.; Person, M.D.; Chao, H.; Tang, D.G.; Wood, R.D. FAM 35A Associates with REV 7 and Modulates DNA Damage Responses of Normal and BRCA 1-defective Cells. EMBO J. 2018, 37, e99543.
- Xu, G.; Chapman, J.R.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 Counteracts DNA Double-Strand Break Resection and Affects PARP Inhibition. Nature 2015, 521, 541–544.
- Dungrawala, H.; Bhat, K.P.; Le Meur, R.; Chazin, W.J.; Ding, X.; Sharan, S.K.; Wessel, S.R.; Sathe, A.A.; Zhao, R.; Cortez, D. RADX Promotes Genome Stability and Modulates Chemosensitivity by Regulating RAD51 at Replication Forks. Mol. Cell 2017, 67, 374–386.e5.
- Rondinelli, B.; Gogola, E.; Yücel, H.; Duarte, A.A.; Van De Ven, M.; Van Der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi, R.; Rottenberg, S.; et al. EZH2 Promotes Degradation of Stalled Replication Forks by Recruiting MUS81 through Histone H3 Trimethylation. Nat. Cell Biol. 2017, 19, 1371–1378.
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.-E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication Fork Stability Confers Chemoresistance in BRCA-Deficient Cells. Nature 2016, 535, 382–387.
- Murai, J.; Feng, Y.; Yu, G.K.; Ru, Y.; Tang, S.-W.; Shen, Y.; Pommier, Y. Resistance to PARP Inhibitors by SLFN11 Inactivation Can Be Overcome by ATR Inhibition. Oncotarget 2016, 7, 76534–76550.
- Soldatenkov, V.A.; Albor, A.; Patel, B.K.; Dreszer, R.; Dritschilo, A.; Notario, V. Regulation of the Human Poly(ADP-Ribose) Polymerase Promoter by the ETS Transcription Factor. Oncogene 1999, 18, 3954–3962.
- Soldatenkov, V.A.; Trofimova, I.N.; Rouzaut, A.; McDermott, F.; Dritschilo, A.; Notario, V. Differential Regulation of the Response to DNA Damage in Ewing’s Sarcoma Cells by ETS1 and EWS/FLI-1. Oncogene 2002, 21, 2890–2895.
- Li, D.; Bi, F.-F.; Cao, J.-M.; Cao, C.; Li, C.-Y.; Liu, B.; Yang, Q. Poly (ADP-Ribose) Polymerase 1 Transcriptional Regulation: A Novel Crosstalk between Histone Modification H3K9ac and ETS1 Motif Hypomethylation in BRCA1-Mutated Ovarian Cancer. Oncotarget 2014, 5, 291–297.
- Molloy-Simard, V.; St-Laurent, J.-F.; Vigneault, F.; Gaudreault, M.; Dargis, N.; Guérin, M.-C.; Leclerc, S.; Morcos, M.; Black, D.; Molgat, Y.; et al. Altered Expression of the Poly(ADP-Ribosyl)Ation Enzymes in Uveal Melanoma and Regulation of PARG Gene Expression by the Transcription Factor ERM. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6219–6231.
- Choul-li, S.; Legrand, A.J.; Bidon, B.; Vicogne, D.; Villeret, V.; Aumercier, M. Ets-1 Interacts through a Similar Binding Interface with Ku70 and Poly (ADP-Ribose) Polymerase-1. Biosci. Biotechnol. Biochem. 2018, 82, 1753–1759.
- Cohen-Armon, M.; Visochek, L.; Rozensal, D.; Kalal, A.; Geistrikh, I.; Klein, R.; Bendetz-Nezer, S.; Yao, Z.; Seger, R. DNA-Independent PARP-1 Activation by Phosphorylated ERK2 Increases Elk1 Activity: A Link to Histone Acetylation. Mol. Cell 2007, 25, 297–308.
- Han, S.; Brenner, J.C.; Sabolch, A.; Jackson, W.; Speers, C.; Wilder-Romans, K.; Knudsen, K.E.; Lawrence, T.S.; Chinnaiyan, A.M.; Feng, F.Y. Targeted Radiosensitization of ETS Fusion-Positive Prostate Cancer through PARP1 Inhibition. Neoplasia 2013, 15, 1207-IN36.
- Lovejoy, C.A.; Xu, X.; Bansbach, C.E.; Glick, G.G.; Zhao, R.; Ye, F.; Sirbu, B.M.; Titus, L.C.; Shyr, Y.; Cortez, D. Functional Genomic Screens Identify CINP as a Genome Maintenance Protein. Proc. Natl. Acad. Sci. USA. 2009, 106, 19304–19309.