Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
Non-alcoholic fatty liver disease (NAFLD) is a clinical–pathological syndrome characterized by excessive deposition of fat and fatty degeneration in liver cells in the absence of excessive alcohol consumption or other known causes of liver disease. The formation of disulfide bonds is a reversible process that can be achieved through thiol–disulfide interchange.
- thioredoxin
- reactive oxygen species
- NAFLD
- gut microbiota dysbiosis
1. Introduction
Non-alcoholic fatty liver disease (NAFLD) is a clinical–pathological syndrome characterized by excessive deposition of fat and fatty degeneration in liver cells in the absence of excessive alcohol consumption or other known causes of liver disease. Pathologically, NAFLD is generally categorized into two types: simple fatty liver and non-alcoholic steatohepatitis (NASH) and can progress to fatty liver fibrosis and even liver cancer. Besides the direct harm from liver disease, NAFLD patients are at significantly increased risk of developing cardiovascular diseases, type 2 diabetes, and chronic renal insufficiency [1,2]. However, the specific pathogenesis of NAFLD/NASH remains unclear, and there is currently no specific treatment available. Clinical pharmacological research on NAFLD is actively underway.
NAFLD is a disease closely related to various factors, including genetics, environment, and diet. To emphasize its metabolic nature, in March 2020, an international expert group proposed renaming it as Metabolic Associated Fatty Liver Disease (MAFLD) [3]. When NAFLD progresses to NASH, the risk of liver fibrosis and liver cancer increases significantly. Consequently, in the natural development of NAFLD, the progression of the disease can be effectively stopped by improving NASH. The exact cause of NAFLD/NASH is still unknown, and the “two-hit” hypothesis proposes that insulin resistance in peripheral adipose tissue results in the breakdown of fats, elevated levels of free fatty acids in the bloodstream, and excessive transportation of fatty acids to the liver, ultimately leading to hepatic steatosis [4,5]. This is the “first hit”. Prolonged and excessive fat accumulation in the liver cannot be eliminated, causing mitochondrial dysfunction. This dysfunction leads to endoplasmic reticulum stress, oxidative stress, and the release of inflammatory factors, which exacerbate hepatocellular damage and contribute to the development of NASH from simple fatty liver disease, referred to as the “second hit”. Reduced secretion of adiponectin (ADPN) and elevated pro-inflammatory factors in peripheral adipose tissue further promote inflammatory responses and worsen insulin resistance, creating a vicious cycle [4]. Additionally, liver cell apoptosis is critical for the development of NASH. The pathogenesis of NAFLD/NASH is highly complex, and the current understanding has shifted from the “two-hit” to the “multiple parallel hits” theory, involving diverse factors such as oxidative stress, gut microbiota, insulin resistance, adipokines, genetics, and epigenetics, and others [5,6].
2. Disruption of Thiol Homeostasis in NAFLD
2.1. Sites of ROS Production
Reactive oxygen species (ROS) are inevitable byproducts of aerobic metabolism in living organisms, including superoxide (O2−), hydrogen peroxide (H2O2), hydroxyl radicals (HO·), and so on. Among them, hydroxyl radicals are the most reactive, capable of attacking almost all biological molecules, inducing lipid peroxidation, and causing DNA double-strand breaks, making them the most destructive type of ROS [7,8], Superoxide (O2−) serves as the precursor of other ROS. When O2 leaks from the mitochondrial respiratory chain, an O2 molecule accepting an electron generates O2−, which can be dismutated by superoxide dismutase (SOD) into H2O2 and O2. H2O2 is not a free radical molecule but can be catalyzed to generate highly active HO· through the Fenton reaction when accepting an electron. The majority of ROS in cells originate from this process [9,10]. The excess ROS causes damage to mitochondrial DNA (mtDNA), respiratory chain complexes, lipids, and other components [11], which, in turn, further induces ROS production, creating a vicious cycle [12,13].
The other source sites of ROS include the endoplasmic reticulum (ER), a crucial site for protein synthesis, post-translational modification, processing, and folding in cells. In the ER lumen, the correct folding of most proteins requires the formation of disulfide bonds between cysteine residues to stabilize their structures. The formation of disulfide bonds is a reversible process that can be achieved through thiol–disulfide interchange. In eukaryotic cells, the folding of oxidative proteins is mainly catalyzed by a series of redox enzymes, such as protein disulfide isomerase (PDI), a member of the thioredoxin superfamily proteins [14]. Additionally, the folding of ER proteins is also dynamically regulated by the balance of the redox buffer, such as GSH/GSSG, which maintains the cellular redox state in a reduced state, GSH accounts for approximately 99% of the overall content, whereas it is found in only 50–60% in the endoplasmic reticulum [15]. PDI transfers two electrons to its substrate ERO1 (ER oxidoreductase 1), producing hydrogen peroxide. This electron transfer mode indicates the induction of ROS production [16].
Moreover, peroxisome, the organelles present in all eukaryotic cells, is believed to be the site to produce ROS. Peroxisome is closely related to the regulation of liver lipid homeostasis [17]. Medium- and long-chain fatty acids (FAs) are oxidized predominantly in mitochondria, whereas very long-chain FAs (VLCFAs) are metabolized almost exclusively by β-oxidation in the peroxisome and are involved in α-oxidation of fatty acids [18]. Peroxisomes produce acetyl coenzyme A through fatty acid β-oxidation, which plays a role in lipid signal transduction to promote lipid autophagy [19]. It is estimated that peroxisomes contribute about 35% of ROS in cells.
Moreover, NADPH oxidase, also known as NOX, is the only enzyme whose main function is to produce ROS. Researchers have identified seven isoforms, NOX1, NOX2, NOX3, NOX4, NOX5, DUOX1, and DUOX2, of which NOX2 is highly expressed in the liver [20]. NOX2 consists of 6 subunits: gp91phox, p22phox, p47phox, p67phox, p40phox, and Rac. Activation of NOX2 requires the translocation of p47phox, p67phox, p40phox, and Rac from the cytoplasm to the cell membrane, where they bind to gp91phox and p22phox to form the complex to produce the oxidants [21]. NOX2 affects signal transduction and immune function by transferring electrons from NADPH to molecular oxygen and generating large amounts of oxidants, a process involved in a variety of physiological activities such as cell proliferation and differentiation [22]. Studies have reported that in liver-resident Kupffer cells and infiltrating macrophages, NOX2-derived ROS stimulate them to produce pro-inflammatory cytokines such as TNF-α, IL-6, and IL-1β in response to a variety of factors, including oxidized low-density lipoproteins (LDL) and lipopolysaccharides (LPS) [23,24,25]. Therefore, over-activation of NOX2 leads to a surge of oxidants, which directly activates downstream inflammatory pathways and indirectly damages DNA and regulates protein phosphorylation, causing oxidative damage and exacerbating inflammation, which leads to cellular dysfunction and the onset and deterioration of diseases and affects the function and metabolism of tissues and organs [26,27]. Moreover, increased NOX2 activity has been shown in patients with NAFLD [28,29], suggesting a correlation between NOX2 and NAFLD. As mentioned above, oxidative stress serves as the “second hit” in the pathogenesis of NAFLD and is typically accompanied by a large amount of ROS. ROS can lead to the inactivation of mitochondrial respiratory chain enzymes and GAPDH, inhibit Ca2+ channels on the membrane, and induce liver cell damage [30]. Furthermore, ROS can induce the production of cytokines and Fas, along with their ligands, exacerbating lipid peroxidation, thus aggravating liver inflammation and hepatocyte fibrosis [31]. Cellular FFA loading can lead to changes in mitochondrial adaptability, enhancing fatty acid oxidation and upregulating electron transport, further resulting in excessive ROS production [32]. Thus, the excessive production of ROS can damage mitochondrial proteins, lipids, and DNA, a process that may play a role in the initiation of NASH. In the late stage of NASH, mitochondrial ultrastructure damage affects its function, reduces ATP synthesis, and intensifies ROS overproduction [33].
2.2. Thioredoxin (Trx) System
When the liver is exposed to high levels of ROS or electrophilic reagents, oxidative damage is prone to occur. At this point, the body initiates a series of antioxidant defense mechanisms [34]. The mammalian thiol-dependent antioxidant system comprises the thioredoxin (Trx) system and the GSH-glutaredoxin (Grx) system, both of which undergo interconversion between thiol (-SH) and disulfide bond (-S-S-) forms to eliminate ROS and maintain the redox homeostasis in the body [35].
The Trx system mainly consists of Trx, thioredoxin reductase (TrxR), and nicotinamide adenine dinucleotide phosphate (NADPH). It is a major thiol-dependent antioxidant system in the body, responsible for maintaining intracellular redox homeostasis and protecting cells from oxidative stress (Figure 1) [9,36]. Mammals have two isoforms of Trxs. Trx1 is present in the cytoplasm with five cysteines, two active site cysteines, and three structural cysteines, while Trx2 is located in the mitochondria with only two cysteine residues in its active center [36]. The Trx system transfers electrons to various key cellular proteins, including peroxiredoxin (Prx), which directly scavenges H2O2, and methionine-S-sulfoxide reductase (MsrA), which participates in oxidative stress
defense [35,37,38]. The proper functioning of the Trx system relies on TrxR accepting electrons from NADPH and transferring them to Trx. The activity and expression levels of TrxR directly determine the normal biological function of Trx and its downstream proteins. Mammalian cells have two forms of TrxR, cytoplasmic TrxR1 and mitochondrial TrxR2, both of which have FAD and NADPH binding domains as well as interface domains. At their N-terminus, they have an active site CVNVGC, and at their C-terminus, they possess a special sequence Gly-Cys-Sec-Gly (GCUG), with Sec being the catalytic active site of TrxR, which is necessary for its reduction activity [39].
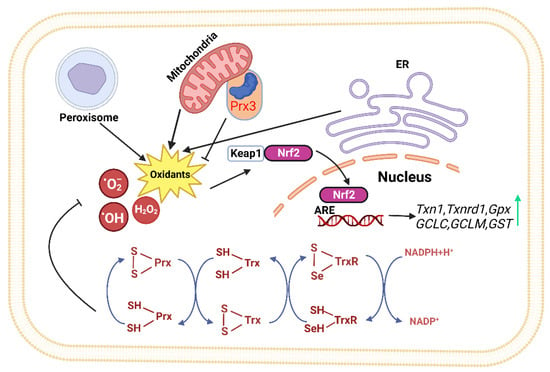
Figure 1. The thioredoxin system plays a crucial role in modulating cell viability and proliferation. Thioredoxin can donate electrons to various enzymes, including peroxiredoxins, which have critical roles in cell signaling by either removing hydrogen peroxide or regulating redox-sensitive signaling molecules [40,41,42,43,44,45]. The redox state of thioredoxin can affect the function of several transcription factors, making it an important player in cellular signaling [46,47,48,49]. (ER: Endoplasmic reticulum, Keap1: kelch like ECH associated protein 1, Nrf2: Nuclear factor erythroid 2-related factor 2, ARE: Antioxidant response element, TrxR: Thioredoxin Reductase, Trx: Thioredoxin, Prx: Peroxiredoxin, NADPH: Nicotinamide Adenine Dinucleotide Phosphate).
Studies have shown significant changes in the Trx system in metabolic syndrome [50,51]. For example, in metabolic syndrome patients, consuming a diet rich in high-saturated fatty acids (HSFA) significantly increased the mRNA levels of Trx in adipose tissue. Simultaneously, postprandial adipose tissue TrxR1 mRNA significantly decreased.This suggests that the ability of Trx to revert from its oxidized form to its reduced form decreased, leading to a compensatory increase in Trx gene levels, indicating increased oxidative stress due to saturated fat intake [52]. The Trx system also plays a crucial role in adipocyte dysfunction and obesity [53]. In obese individuals with metabolic disorders, TrxR activity and Trx content in subcutaneous tissue are significantly increased because subcutaneous fat seems to provide good protection against increased oxidation in obese subjects compared to visceral fat. Protein levels of Trx, GPx, and CuZnSOD, as well as activities of GPx, GR, GST, TrxR, and SOD, were significantly higher in “at-risk” obese women than in metabolically healthy women [54]. While the expression of Trx-dependent peroxiredoxin (Prx3) in adipose tissue is significantly reduced [55], Prx3 is a key molecule regulating adipocyte oxidative stress and adipokine expression [56]. This may indicate that expression of various antioxidant enzymes is predominantly and specifically regulated by different transcription factors, but the detailed mechanism behind it needs further clarification.
Trx system in NASH has been shown to be changed in several studies [57,58]. Serum Trx levels are significantly increased in NASH patients compared to simple steatosis patients, making it a potential biomarker to distinguish NASH from early-stage fatty liver [59]. In animal models of choline-deficient liver steatosis, the activities of hepatic Trx1 and TrxR are upregulated at day 14 but significantly decreased at day 30 compared to day 14 [60]. Furthermore, the level of thioredoxin-interacting protein (TXNIP) in the liver of NAFLD patients is significantly elevated [61], and in mice fed a methionine and choline-deficient diet to induce NASH, the TXNIP gene was overexpressed, and expression of hepatic TrxR1 and TrxR2 decreased [62].
2.3. Glutathione (GSH)-Grx System
In addition to the Trx system, the glutathione-glutaredoxin (GSH-Grx) system is another crucial thiol-dependent redox system involved in cellular redox balance regulation [63]. It comprises NADPH, glutathione reductase (GR), and GSH coupled with Grxs, including cytosolic Grx1 and mitochondrial Grx2. The GSH-Grx systems mainly mediate redox signal transduction by reversibly catalyzing protein S-glutathionylation. GSH is the most abundant thiol small molecule, playing a vital role in maintaining intracellular redox homeostasis [64]. GSH can reduce various proteins, including glutathione peroxidases and glutathione transferases [65], and the resulting GSSG can be recycled by GR using NADPH as an electron donor [66].
Studies have shown that when Glrx1 KO mice are given a standard diet for some time, they develop obesity, hyperlipidemia, and fatty liver, resembling NAFLD. When fed a high-fat diet, Glrx1 KO mice develop fatty liver inflammation and progress to NASH [67]. In NAFLD animal models fed with a high-fat diet, S-glutathionylated protein levels increase, and an important target protein, Sirtuin-1 (SirT1), is identified. SirT1 is a NAD+-dependent class III histone deacetylase that regulates key transcription factors coordinating hepatic lipid metabolism [68,69,70]. Activation of SirT1 ameliorates Non-Alcoholic Fatty Liver (NAFL); conversely, hepatic SirT1 deficiency leads to steatosis [71], and thiol modification of SirT1 regulates lipid metabolism through acetylation of key transcription factors. S-glutathionylation inactivates SirT1 and promotes hyperacetylation and activation of downstream target proteins such as p53 and sterol regulatory element binding protein (SREBP) [68]. Activation of p53 is a cellular response to stress, resulting in the alteration of metabolism and the arrest of the cell cycle, and severe oxidative damage to hepatocytes may trigger p53 to induce cell death [72]. Studies also suggest that SirT1 overexpression can improve NAFLD [73], while Sirt1 knockout in mice leads to NAFLD (Figure 2) [74].
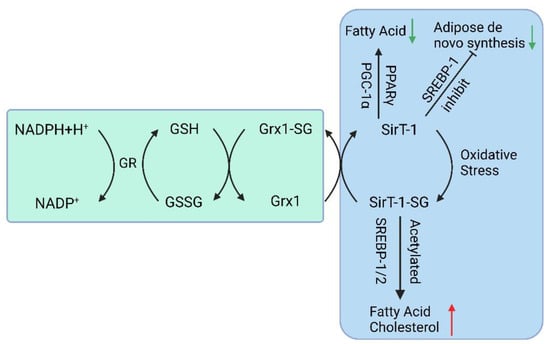
Figure 2. The GSH-Grx 1 system controls protein S-glutathionylation major players in the NAFLD. (GR: Glutathione Reductase, GSH: Glutathione, NADPH: Nicotinamide Adenine Dinucleotide Phosphate, Grx: Glutaredoxin, SirT-1: sirtuin type 1, PPAR: Peroxisome proliferators-activated receptors, PGC-1α: Peroxisome proliferator-activated receptor-gamma coactivator, SREBP-1c: Sterol regulatory element binding protein-1c).
Grx2 has two known isoforms encoded by the same gene (GLRX2): Grx2a (mitochondrial) and Grx2c (cytoplasmic) [75,76]. The role and effects of mitochondrial Grx2-mediated glutathionylation in cellular metabolism are still poorly understood. A mouse model with a depletion of mitochondrial Grx2 fed with a standard diet had spontaneous weight gain and accumulation of lipid droplets in the liver, suggesting that Grx2 is involved in mitochondrial redox environment and lipid metabolism regulation [77]. Genes such as cytochrome P-450 7a1 (Cyp7a1), which is specifically expressed in the liver and participates in the reduction of cholesterol, were found to be significantly downregulated in the mice model [77].
2.4. Nrf2 Signaling Pathway
The Keap1-Nrf2-ARE pathway is one of the major pathways that maintain cellular homeostasis during oxidative stress in the liver and is also regulated by Trx. Its function is to protect cells from endogenous and exogenous damage caused by oxidative stress. When Nrf2 accumulates significantly in the cell nucleus, a large number of antioxidant and phase II detoxification enzyme genes are upregulated [78]. Previous studies have identified over 200 downstream target genes regulated by Nrf2 in human cells [79], including the genes encoding NAD(P)H quinone oxidoreductase 1 (NQO1), HO-1, GST, GPx, CAT, SOD, and GR (Figure 1) [80]; It controls the expression of the glutamate–cysteine ligase catalytic subunit (GCLC) and glutamate–cysteine ligase modifier subunit (GCLM) to maintain the proper ratio of GSH to GSSG in cells [81]; Nrf2 also positively regulates some enzymes involved in liver detoxification, including Trx1, TrxR1, Gpx2, and GST [82,83]. These detoxification enzymes can eliminate H2O2, free radicals, and oxidized thiols in the cytoplasm, endoplasmic reticulum, and mitochondria [84]. Thus, Nrf2 represents a potential therapeutic target for NAFLD.
In addition to Nrf2, which has been shown to bind to the ARE of the Trx, TrxR, and Prx1 promoters [85,86,87], hypoxia-inducible factor (HIF)-1α synergistically up-regulates specific genes during hypoxia with another class of transcription factors, the E26 translationally specific (Ets) family of related proteins [88]. In human PC3 prostate cancer cells, the Prx1 gene promoter was significantly induced by either H2O2 or reoxygenation after hypoxic growth [89]. Co-transfection of constructs overexpressing Ets-1 or Ets-2 with the Prx1 promoter construct resulted in increased promoter activity, and ChIP assays also confirmed that both Ets-1 and Ets-2 bound to the Prx1 gene promoter in PC3 cells [89]. Thus, it can be hypothesized that the Ets pathway may play a role in regulating redox control during hypoxia and reoxygenation to complement the activation of the Nrf2 pathway. In addition, specificity protein 1 (Sp1), Fas/Jun, TATA box binding protein (TBP), cAMP response element binding protein (CREB), retinoic acid receptor/retinoid X receptor (RAR/RXR), and other transcription factors bind to their DNA sites to regulate Trx [85,87,90,91]. Retinoid X receptor (RAR/RXR) and other transcription factors that bind to DNA sites to regulate Trx; Octamer binding protein (Oct-1), Sp1, Sp3, and other transcription factors bind to the DNA site to regulate TrxR [92]. However, whether these transcriptional regulations are involved in NAFLD/NASH is unclear.
This entry is adapted from the peer-reviewed paper 10.3390/antiox12091680
This entry is offline, you can click here to edit this entry!