Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Pathology
The liver is an organ that is particularly exposed to reactive oxygen species (ROS), which not only arise during metabolic functions but also during the biotransformation of xenobiotics. The disruption of redox balance causes oxidative stress, which affects liver function, modulates inflammatory pathways and contributes to disease.
- ROS
- liver disease
- fibrosis
- hepatitis
1. Major Types of Liver Cells and Their Role in Liver Functions
The liver is a large organ constituting about 2% of body weight in adult humans [1]. It is anatomically divided into a larger right and a smaller left lobe, each made up of thousands of lobules. The liver lobules contain parenchymal and non-parenchymal cells that interact to form a functional hepatic unit (Figure 1) [2]. Parenchymal cells include hepatocytes, the predominant liver cell population, and cholangiocytes. Non-parenchymal cells include sinusoidal endothelial cells, macrophages, stellate cells and natural killer cells [3,4].
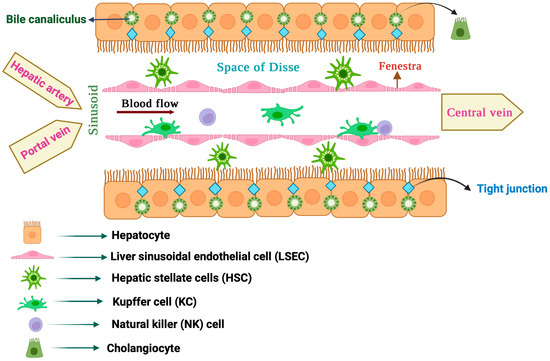
Figure 1. Anatomy of the liver lobule.
1.1. Hepatocytes
The liver parenchyma is mostly composed of hepatocytes, which make up to 80% of total liver cells [5]. They perform vital functions such as the clearance of toxic metabolites and xenobiotics, as well as the secretion of proteins and lipids to maintain blood homeostasis. Hepatocytes also produce hormones [6] and bile [7], and mediate innate immune responses [8]. Nutrient-rich blood from the portal vein and oxygenated blood from the hepatic artery are directed to hepatocytes via highly specialized capillaries known as hepatic sinusoids [9]. In liver lobules, hepatocytes are arranged into plates around the central vein, which are separated by liver sinusoids. To enhance absorption from the plasma, hepatocytes extend a number of microvilli into the space of Disse, a thin peri-sinusoidal region between the hepatocytes and sinusoidal endothelium [10].
1.2. Cholangiocytes
Cholangiocytes are specialized epithelial cells lining the extra and intrahepatic bile ducts, which make up 3–5% of liver cells [11]. They are involved in the synthesis (together with hepatocytes), secretion and modification of bile, which is essential for the digestion and absorption of fats. Cholangiocytes also regulate bile flow in response to hormonal and neural signals and exhibit immune functions. They represent a heterogeneous cell population of biochemically and morphologically distinct large and small cholangiocytes.
1.3. Liver Sinusoidal Endothelial Cells (LSECs)
LSECs are the most abundant type of liver non-parenchymal cells, constituting about 15–20% of the total liver cell content [12]. They are very specialized endothelial cells that discontinuously line the wall of hepatic sinusoids and make a unique permeable barrier between blood within the sinusoids and the underlying hepatocytes and hepatic stellate cells. LSECs have distinctive morphological features, including the presence of numerous small open pores called fenestrae, and the absence of a classical basement membrane and diaphragm, which help their permeability. LSEC fenestration provides access of hepatocytes to certain substances from the circulating blood by connecting the lumen of the sinusoids with the space of Disse. In addition, LSECs have important physiological and immunological functions such as the regulation of hepatic vascular tone by synthesizing nitric oxide (NO) and endothelin-1 (ET-1), the maintenance of hepatic stellate cells and Kupffer cells in a quiescence state, filtration, endocytosis, antigen presentation and leukocyte recruitment. LSECs are the first liver cells to be affected by liver injury and contribute to the initiation and progression of liver disease. Thus, following liver injury, LSECs undergo morphological changes culminating in capillarization (defenestration), which causes sinusoidal endothelial dysfunction [13,14].
1.4. Hepatic Stellate Cells (HSCs)
HSCs, also known as Ito cells, are non-parenchymal liver pericytes located in the space of Disse [15,16]. They store 50–95% of the body’s vitamin A and can communicate with all cell types within the liver, either physically or through cytokines and chemokines. HSCs are considered liver-specific mesenchymal stem cells (MSCs), as they possess properties akin to stem/progenitor cells. In the healthy liver, quiescent HSCs play a vital role in vitamin A homeostasis. They also contribute to the regulation of extracellular matrix (ECM) turnover, immunoregulation, expression of growth factors required for liver development and regulation of hepatic blood flow with their contractile ability. Moreover, HSCs are critical for liver regeneration and repair [17]. In response to liver injury, quiescent vitamin A-rich HSCs undergo activation or trans-differentiation into proliferative and contractile myofibroblast-like cells, which is a key event in liver fibrosis [18]. Activated HSCs lose their vitamin A content and express α-smooth muscle actin (α-SMA; a key HSC activation marker), while they synthesize and release specific ECM components, such as collagens, proteoglycans and glycoproteins [18].
1.5. Liver Macrophages
Liver resident macrophages, also known as Kupffer cells (KCs), are located in the lumen of sinusoids adherent to LSECs [19]. They constitute the largest population of mononuclear phagocytes in the body and are considered a filter for gut-derived pathogens within the portal circulation. In physiological states, KCs contribute to immune surveillance by eliminating circulating pathogens and hazardous materials via phagocytosis. In addition, KCs play a key role in systemic iron homeostasis by clearing senescent red blood cells via erythrophagocytosis, which is coupled with the recycling of iron to the bloodstream for de novo erythropoiesis [20]. KCs are also crucial for cholesterol and lipid metabolism. Liver macrophages were earlier considered to consist of a single population of KCs. However, the use of advanced single-cell and spatial transcriptomics technologies uncovered an unexpected heterogeneity of liver macrophage populations [21]. Under pathological conditions, KCs and other liver residents, as well as recruited macrophages, are activated through various inducers and can acquire distinct phenotypes that affect liver disease outcomes [22].
1.6. Liver Natural Killer (NK) Cells
Liver resident NK cells, also known as Pit cells, are primarily found in the sinusoidal spaces. They are strategically positioned to monitor and respond to various substances, including pathogens and tumor cells, that enter the liver through the bloodstream. Pit cells exhibit morphological features of large granular lymphocytes and differ from circulating NK cells in several aspects. For instance, they are physically connected with their pseudopodia to the microvilli of hepatocytes in the space of Disse. Pit cells are a major part of the hepatic innate immune system, participate in host defense mechanisms against tumors and microbes, and are critical for maintaining immune balance [23].
2. Oxidative Stress in the Liver
Reactive oxygen species (ROS) such as superoxide radical (O2•−) and hydrogen peroxide (H2O2) emerge from the incomplete reduction of molecular oxygen. They are physiologically produced in mitochondria during aerobic respiration and are also generated in cells via enzymatic reactions. H2O2 can oxidize ferrous (Fe2+) to ferric (Fe3+) iron according to Fenton chemistry, which yields the short-lived but highly toxic hydroxyl radical (OH•) [24]. Ferric iron undergoes redox cycling and is subsequently reduced to ferrous by O2•−. Thus, in the presence of O2•− and H2O2, iron acts as a catalyst for the generation of OH•, which may give rise to other reactive free radicals and propagate lipid peroxidation. On the other hand, O2•− can react with nitrogen monoxide radical (NO•) to yield peroxynitrite (NOO−), a potent oxidant. Reactive free radicals and non-radical oxidants can attack and damage all cellular macromolecules, including proteins, nucleic acids and lipids. Protein oxidation may have negative functional consequences, while oxidation of nucleic acids may lead to mutagenesis. Peroxidation of membrane lipids, especially polyunsaturated fatty acids (PUFAs), may promote ferroptosis, an iron-dependent form of cell death [25].
ROS such as O2•− and OH• are unstable free radicals. They owe their reactivity to their capacity to extract an electron from, or donate their unpaired electron to neighboring molecules, and thereby acquire a thermodynamically stable state. By contrast, H2O2 or NOO− are stable non-radical oxidants. While ROS were initially considered as biohazards, it is now clear that at low levels they also act as second messengers and play a major role in physiological signaling pathways, gene expression regulation, host defense against microorganisms, immune responses and vasodilation. However, an uncontrolled rise of ROS levels is toxic and promotes “oxidative stress”, which has been defined as a disruption of the oxidant/antioxidant balance in favor of the former that can lead to tissue damage [26]. This pathologic state is lately also referred to as “oxidative distress”, to distinguish it from homeostatic “oxidative eustress” [27]. The same concept applies to NO• and other reactive nitrogen species (RNS): at low levels, they act as important second messengers, while in excess they disrupt the redox balance causing “nitrosative stress” [28].
The liver is an important site of ROS production by virtue of its metabolic and detoxification activities. ROS are generated via the mitochondrial respiratory chain and from other sources including peroxisomes, xanthine oxidases, cytochrome P450 oxidases and NADPH oxidases (NOXs) (Figure 2) [29]. Oxidases of the cytochrome P450 (CYP) family, such as ethanol-induced cytochrome P450 2E1 (CYP2E1), are involved in xenobiotics metabolism and constitute major sources of ROS in hepatocytes. Oxidases of the NOX family utilize NADPH and molecular oxygen to produce O2•−, which is rapidly dismutated to H2O2. The NOX2 isoform was originally characterized in phagocytes but is also expressed in KCs. Hepatocytes, LSECs and HSCs express various isoforms including NOX2 and the non-phagocytic NOX1, NOX4 and NOX5, as well as the dual oxidases DUOX1 and DUOX2 [30,31]. NOX4 has a unique mode of action compared to other NADPH oxidases and is known to predominantly generate H2O2 instead of O2•− [32].
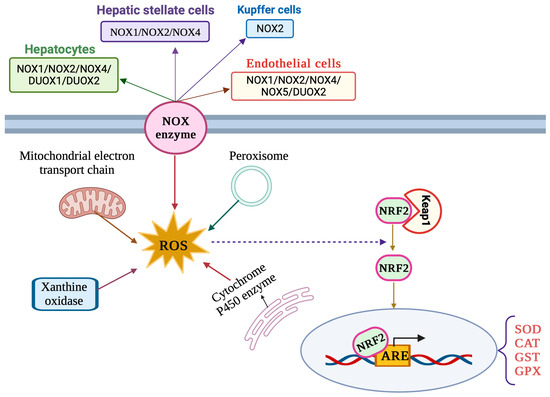
Figure 2. Major sources of ROS production and NRF2 signaling pathway in liver cells.
3. Antioxidant Defense Mechanisms in the Liver
Liver cells possess robust antioxidant defense mechanisms consisting of enzymatic and non-enzymatic components, which enable them to keep ROS at physiological levels. Antioxidant enzymes include superoxide dismutases (SOD), cytosolic glutathione peroxidases (GPX), glutathione reductases (GRX), peroxiredoxins (PRX), thioredoxins (TRX) and catalase (CAT). Glutathione (GSH), bilirubin, ubiquinone (coenzyme Q10), uric acid and vitamins E, A and C are established non-enzymatic antioxidants [33]. Dietary antioxidants, such as curcumin, resveratrol, quercetin and other flavonoids are also thought to contribute to protection against oxidative stress [34].
In general, enzymatic antioxidants metabolize ROS, while non-enzymatic antioxidants prevent or attenuate oxidative damages by neutralizing free radicals and non-radical oxidants. Inhibition of the activity or expression of enzymes involved in free radical production, such as NOXs and xanthine oxidases, or increasing the activity or expression of intracellular antioxidant enzymes is also part of the antioxidant defense armamentarium [35].
The NRF2/ARE signaling pathway (Figure 2) is considered the main cellular antioxidant defense mechanism [36] and has critical functions in liver pathophysiology [37]. NRF2 (nuclear factor erythroid 2-related factor 2) is a member of the Cap’n’Collar (CNC) basic leucine zipper (bZIP) family of transcription factors. Its primary function is to regulate the expression of a wide array of antioxidant and detoxification genes. Under normal physiological conditions, NRF2 is sequestered in the cytoplasm by its inhibitor, Kelch-like ECH-associated protein 1 (Keap1). Keap1 acts as a sensor for oxidative stress and facilitates the degradation of NRF2 via the proteasomal pathway. However, when cells encounter oxidants or electrophilic insults, specific cysteine residues on Keap1 are modified, leading to conformational changes in the Keap1–NRF2 complex. This results in the liberation and nuclear translocation of NRF2. Once in the nucleus, NRF2 forms a heterodimer with small musculoaponeurotic fibrosarcoma (MAF) proteins and binds to antioxidant response elements (AREs) present in the promoter regions of target genes [36].
The binding of NRF2 to AREs initiates the transcription of a battery of cytoprotective genes, including antioxidant enzymes (e.g., SOD, CAT and GPX), phase II detoxifying enzymes (e.g., glutathione S-transferases, NAD(P)H quinone oxidoreductase 1) and other stress response proteins. These gene products contribute to the cellular defense against oxidative stress by enhancing the intracellular antioxidant capacity and by promoting detoxification processes. The NRF2/ARE pathway is operational in parenchymal, as well as non-parenchymal, cells of the liver [37].
4. The Impact of Oxidative Stress on Liver Cells
The liver is continuously exposed to different toxic and reactive metabolites including ROS and RNS. A shift in the redox balance toward oxidative stress can be considered an initial step in the pathogenesis of liver diseases [38]. This process is affected by comorbidities such as diabetes/insulin resistance and by various exogenous factors such as alcohol abuse, viral infection, drug overdose, high-caloric diet, and exposure to environmental toxins, UV light or heavy metals. A surge in ROS and RNS levels is important in the onset of inflammatory reactions, fibrosis, necrosis, apoptosis or malignant transformation [28,38].
Hepatocytes are important sites of ROS production, especially in mitochondria, and are also sensitive to ROS-mediated injury. Each hepatocyte contains 1000 to 2000 mitochondria occupying about 20% of the cell volume [39]. ROS-mediated damage of lipids and particularly, PUFAs, can alter cell membrane fluidity and permeability. Mitochondrial lipid peroxidation negatively affects the electron transport chain, aggravating ROS production and oxidative stress [38]. Mitochondrial dysfunction in hepatocytes has been linked to the development and progression of chronic liver disorders [38,40]. For instance, patients with non-alcoholic steatohepatitis (NASH) exhibit hepatic oxidative stress due to impaired mitochondrial respiratory capacity and proton leakage [41]. In a mouse model of fatty liver disease, pharmacological improvement of mitochondrial redox homeostasis with the flavonoid dihydromyricetin was shown to be hepatoprotective [42]. Hjv−/− mice, a model of hereditary iron overload (hemochromatosis), exhibit mitochondrial hyperactivity in hepatocytes, which predisposes them to HCC [43]. On the other hand, pharmacological chelation of mitochondrial iron has been shown to promote mitophagy, which protects mice against HCC [44]. Experiments with primary rat hepatocytes and rat H4IIEC3 hepatoma cells showed that palmitate treatment promotes a flux of calcium from ER to mitochondria, which causes mitochondrial oxidative stress and lipotoxicity. These data highlight the physiological importance of finetuning mitochondrial activities and redox balance. Accumulation of ROS can induce hepatocellular dysfunction or death that will eventually result in the release of damage-associated molecular patterns (DAMPs). Under these circumstances, non-parenchymal cells, such as KCs, HSCs and newly recruited immune cells are activated and produce pro-fibrogenic and pro-inflammatory mediators [45].
Oxidative stress promotes an influx of calcium into cells and redistribution of cellular calcium from the endoplasmic reticulum (ER) to the cytosol, mitochondria and nuclei, which in turn may trigger apoptotic and necrotic death [46]. These responses increase mitochondrial permeability transition and facilitate the release of pro-apoptotic factors such as cytochrome c, and the activation of calcium-dependent endonucleases, proteases and lipases, contributing to the death of hepatocytes and other liver cell types [47]. In addition, oxidative stress can affect the secretory functions of hepatocytes by disrupting the formation of bile flow, leading to cholestasis [48].
While ROS and lipid peroxidation products impair hepatocellular function and viability, they promote the differentiation and activation of HSCs to myofibroblasts, leading to the secretion and accumulation of collagen and other ECM components within the liver [49]. Therefore, chronic activation of HSCs in response to oxidative stress favors the development of liver fibrosis, which may progress to cirrhosis and HCC [50,51].
KCs are also activated by certain stimulants leading to ROS production, expression of a variety of cytokines and pro-inflammatory mediators, and recruitment of more immune cells [52]. Experimental studies with animal models have shown that ROS originating from KCs play a prominent role in the development of liver injury in response to hepatotoxins [53,54,55]. KCs secrete transforming growth factor β (TGF-β) and platelet-derived growth factor (PDGF), which in turn promote HSC activation, contributing to liver fibrosis [52]. In addition, KCs can directly kill hepatocytes through the activation of Fas-dependent apoptosis [56].
LSECs are sensitive to oxidative stress mainly due to their low H2O2 clearance capacity [57,58], but also due to their exposure to gut-derived toxins carried in the portal vein [59]. Therefore, ROS can selectively damage LSECs and impair their physiological activities. For instance, the oxidation of spectrin can disrupt its interaction with actin, which is essential to maintain fenestrae structure and function, and thereby cause fenestrae closure [60]. Defenestration impairs the bidirectional exchange of molecules between hepatocytes and hepatic blood sinuses [61]. Vascular endothelial dysfunction driven by oxidative stress and inflammation plays an important role in liver injury [62,63,64]. Thus, it may lead to the decreased generation of vasodilator factors such as NO and promote vasoconstriction, which causes increased resistance in sinusoidal microcirculation and portal hypertension [65].
Autophagy is the main endogenous recycling process that preserves cell homeostasis under physiological conditions and offers a survival mechanism under stress [66]. Experimental studies with animal models suggest that autophagy protects LSECs against oxidative stress responses to acute liver injury, while impairment of this pathway leads to endothelial dysfunction and contributes to HSC activation and liver fibrosis [62]. Along these lines, NASH patients exhibit smaller autophagic vacuoles in LSECs [67], indicating that autophagy is dysregulated in liver diseases.
Cholangiocytes are involved in cholangiopathies, which can be mediated by oxidative stress factors [48]. Nevertheless, the effects of oxidative stress on cholangiocyte pathophysiology are not well understood. As an example, increased oxidative stress can induce senescence in cholangiocytes through the stimulation of ER stress [68]. On the other hand, melatonin appears to protect cholangiocytes against oxidative stress-induced cell damage and inflammation [69].
NK cells are generally susceptible to ROS, and oxidative stress can alter their activity. This can contribute to immune escape within the tumor microenvironment [70,71]. However, the role of oxidative stress on liver resident NK cells (Pit cells) in the context of liver disease is not clear. The effects of oxidative stress on the various liver cell types are schematically illustrated in Figure 3.
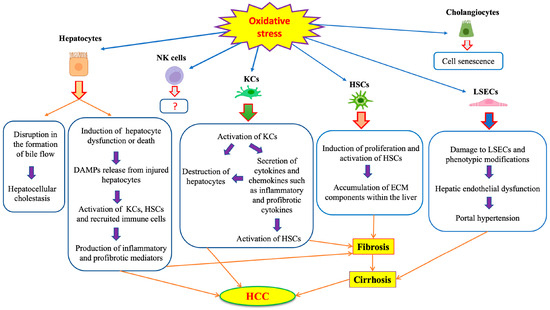
Figure 3. Effects of oxidative stress on different liver cells.
This entry is adapted from the peer-reviewed paper 10.3390/antiox12091653
This entry is offline, you can click here to edit this entry!